分析 Visium HD 数据#
Visium HD 是 10x Genomics 推出的高分辨率空间转录组方案,用于在组织切片上直接测量基因表达。与更早期、分辨率较低的 spot 级方案相比,Visium HD 在分析上的实际变化是:我们往往要从更密集的空间单元出发,再在同一块组织的多个视图之间切换,例如规则 bin 视图和分割后的细胞视图。因此,分析重点不再只是表达矩阵本身,还包括坐标、组织图像和分割结果之间如何彼此对应。
这个教程演示了如何在 OmicVerse 中完成一条典型的 Visium HD 工作流程,同时使用 bin-level output 和 cell segmentation output 两种数据视图。整个示例覆盖四项任务:
将 Visium HD 数据读入
AnnData;在组织图像上可视化基因表达;
在局部区域和全切片范围内识别空间变异基因;
构建低维表示,用于聚类和空间解释。
下面的代码默认你已经把 Visium HD 输出文件下载并整理到 data/visium_hd/ 目录下。
环境设置#
我们先导入 OmicVerse,并启用两个 notebook 中常用的便利功能:
ov.style(font_path='Arial'):为后续图形设置统一的绘图风格;%load_ext autoreload和%autoreload 2:在开发或调试时自动重载修改过的 Python 模块,无需反复重启内核。
import omicverse as ov
ov.style(font_path='Arial')
# 启用自动重载,便于开发和调试
%load_ext autoreload
%autoreload 2
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA A100-SXM4-40GB
Memory: 39.4 GB | Compute: 8.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.10rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
ov.settings.cpu_gpu_mixed_init() 会以 CPU/GPU 混合模式初始化 OmicVerse。对于一部分仍然偏 CPU 的预处理步骤,以及一部分可以从 GPU 加速中受益的数值计算步骤,这种模式通常更稳妥。
ov.settings.cpu_gpu_mixed_init()
CPU-GPU mixed mode activated
Available GPU accelerators: CUDA
加载 Visium HD 数据集#
Visium HD 在非常精细的空间分辨率下提供表达测量结果。实际分析时,同一份数据通常有两种视图都很有价值:
bin-level representation:表达计数存放在规则网格上;
cell-segmentation representation:将 bin 重新分配到分割后的细胞。
本教程会同时演示这两种视图。bin-level 对象更适合检查原始空间信号和组织图像对齐情况,而 segmentation-level 对象则更适合后续以细胞为中心的分析、空间特征发现和聚类。
在正式加载矩阵前,先查看目录结构通常会更稳妥。下面的辅助函数会打印文件树,并跳过 analysis 目录,让输出更聚焦于数据读入真正需要的文件。
这里使用的是 10x Genomics 提供的公开 Visium HD 人前列腺癌 FFPE 示例数据集,计数矩阵和组织图像可从以下页面获取: https://www.10xgenomics.com/datasets/visium-hd-cytassist-gene-expression-libraries-human-prostate-cancer-ffpe
from pathlib import Path
ov.utils.print_tree(
Path("data/visium_hd/binned_outputs/square_016um"),
skip_dirs={"analysis"}
)
square_016um/
spatial/
aligned_fiducials.jpg
detected_tissue_image.jpg
tissue_positions.parquet
aligned_tissue_image.jpg
tissue_lowres_image.png
cytassist_image.tiff
tissue_hires_image.png
scalefactors_json.json
raw_feature_bc_matrix.h5
filtered_feature_bc_matrix/
barcodes.tsv.gz
features.tsv.gz
matrix.mtx.gz
filtered_feature_bc_matrix.h5
raw_feature_bc_matrix/
barcodes.tsv.gz
features.tsv.gz
matrix.mtx.gz
读取 bin-level 输出#
第一个对象来自 square_016um 目录,这里存放的是基于规则网格分箱后的 Visium HD 数据。当你想先检查原始高分辨率空间信号,而不是立刻进入细胞层面解释时,这个视图尤其有用。
ov.io.read_visium_hd() 中有几个参数值得特别关注:
path:所选 Visium HD 输出目录的根路径;data_type='bin':告诉 OmicVerse 将输入解释为 bin-level 矩阵,而不是分割后的细胞;cell_matrix_h5_path和count_mtx_dir:分别指向 HDF5 格式和目录格式的过滤后计数矩阵;tissue_positions_path:提供每个 bin 的空间坐标;hires_image_path、lowres_image_path和scalefactors_path:把分子数据与组织图像关联起来,供后续绘图使用。
adata_hd = ov.io.read_visium_hd(
path="data/visium_hd/binned_outputs/square_016um",
data_type="bin",
cell_matrix_h5_path="filtered_feature_bc_matrix.h5",
count_mtx_dir='filtered_feature_bc_matrix',
tissue_positions_path = "spatial/tissue_positions.parquet",
# if figure and scalefactor stored in outs/spatial
hires_image_path="spatial/tissue_hires_image.png",
lowres_image_path="spatial/tissue_lowres_image.png",
scalefactors_path="spatial/scalefactors_json.json",
)
[VisiumHD][INFO] read_visium_hd entry (data_type='bin')
[VisiumHD][START] Reading bin-level data from: /scratch/users/steorra/analysis/26_omic_protocol/data/visium_hd/binned_outputs/square_016um
[VisiumHD][INFO] Sample key: square_016um
[VisiumHD][STEP] Loading count matrix (h5='filtered_feature_bc_matrix.h5', mtx='filtered_feature_bc_matrix')
[VisiumHD][STEP] Loading tissue positions: /scratch/users/steorra/analysis/26_omic_protocol/data/visium_hd/binned_outputs/square_016um/spatial/tissue_positions.parquet
[VisiumHD][STEP] Loading images and scale factors
[VisiumHD][OK] Done (n_obs=139446, n_vars=18132)
返回结果是一个标准的 AnnData 对象。此时快速检查一下观测数、基因数,以及 obs、var、obsm 和 uns 的内容,会很有帮助。
adata_hd
AnnData object with n_obs × n_vars = 139446 × 18132
obs: 'in_tissue', 'array_row', 'array_col', 'pxl_row_in_fullres', 'pxl_col_in_fullres'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'spatial'
obsm: 'spatial'
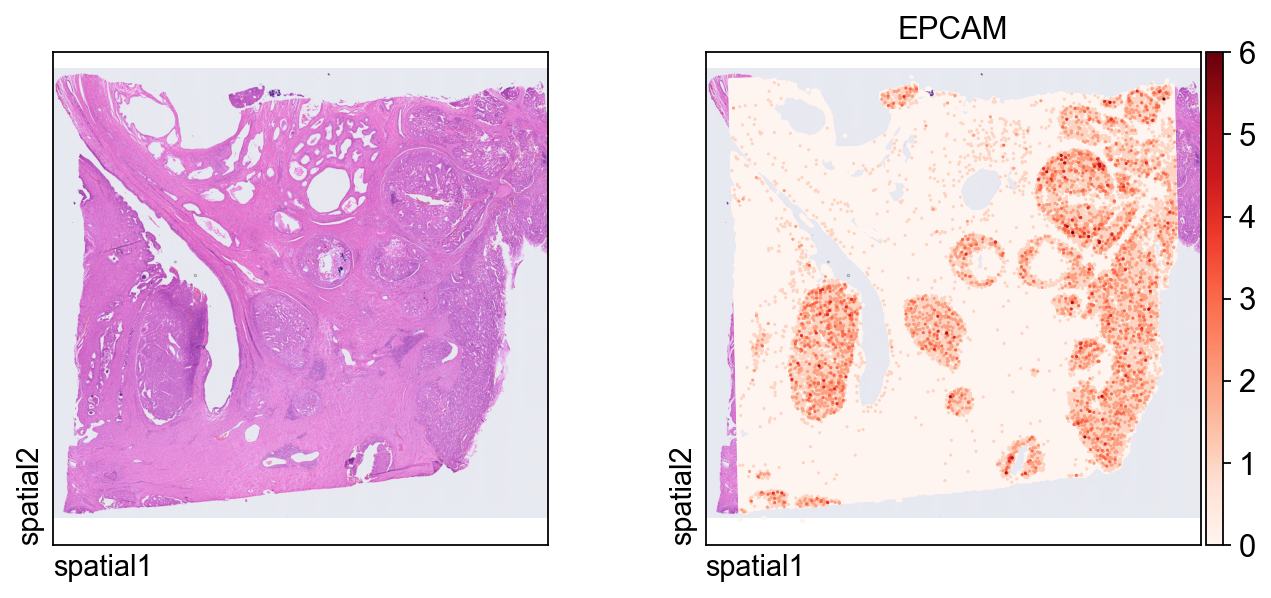
第一张图把表达信号叠加在组织图像上。
几个关键绘图参数:
color=[None, "EPCAM"]:同时显示纯组织图像和EPCAM的表达;size:每个空间单元的点大小;linewidth=0:移除点边框,让高密度绘图更干净;cmap='Reds':对非负表达值使用连续红色配色。
ov.pl.spatial(
adata_hd, color=[None,"EPCAM"],
size=3, linewidth=0,
legend_fontsize=13, frameon=None,
cmap='Reds'
)
读取细胞分割输出#
接下来切换到 segmentation-level 输出,此时每个观测值对应的是分割后的细胞,而不再是规则 bin。这个表示通常更适合细胞层面的可视化、空间特征发现和聚类分析。
相较于 bin-level 导入,主要差别在于这里额外引入了分割文件:
data_type='cellseg':启用细胞分割解析模式;cell_segmentations_path:指向包含细胞 polygon 边界的geojson文件。
这两个对象实际上提供的是同一块组织的互补视图:bin-level 更适合检查原始空间结构,segmentation-level 更适合后续细胞层面的分析。
adata_seg = ov.io.read_visium_hd(
path="data/visium_hd/segmented_outputs",
data_type="cellseg",
cell_matrix_h5_path="filtered_feature_cell_matrix.h5",
cell_segmentations_path="graphclust_annotated_cell_segmentations.geojson",
# if figure and scalefactor stored in outs/spatial
hires_image_path="spatial/tissue_hires_image.png",
lowres_image_path="spatial/tissue_lowres_image.png",
scalefactors_path="spatial/scalefactors_json.json",
)
[VisiumHD][INFO] read_visium_hd entry (data_type='cellseg')
[VisiumHD][START] Reading cell-segmentation data from: /scratch/users/steorra/analysis/26_omic_protocol/data/visium_hd/segmented_outputs
[VisiumHD][INFO] Sample key: segmented_outputs
[VisiumHD][STEP] Loading segmentation geometry: /scratch/users/steorra/analysis/26_omic_protocol/data/visium_hd/segmented_outputs/graphclust_annotated_cell_segmentations.geojson
[VisiumHD][STEP] Loading count matrix: /scratch/users/steorra/analysis/26_omic_protocol/data/visium_hd/segmented_outputs/filtered_feature_cell_matrix.h5
[VisiumHD][STEP] Loading images and scale factors
[VisiumHD][OK] Done (n_obs=227257, n_vars=18132)
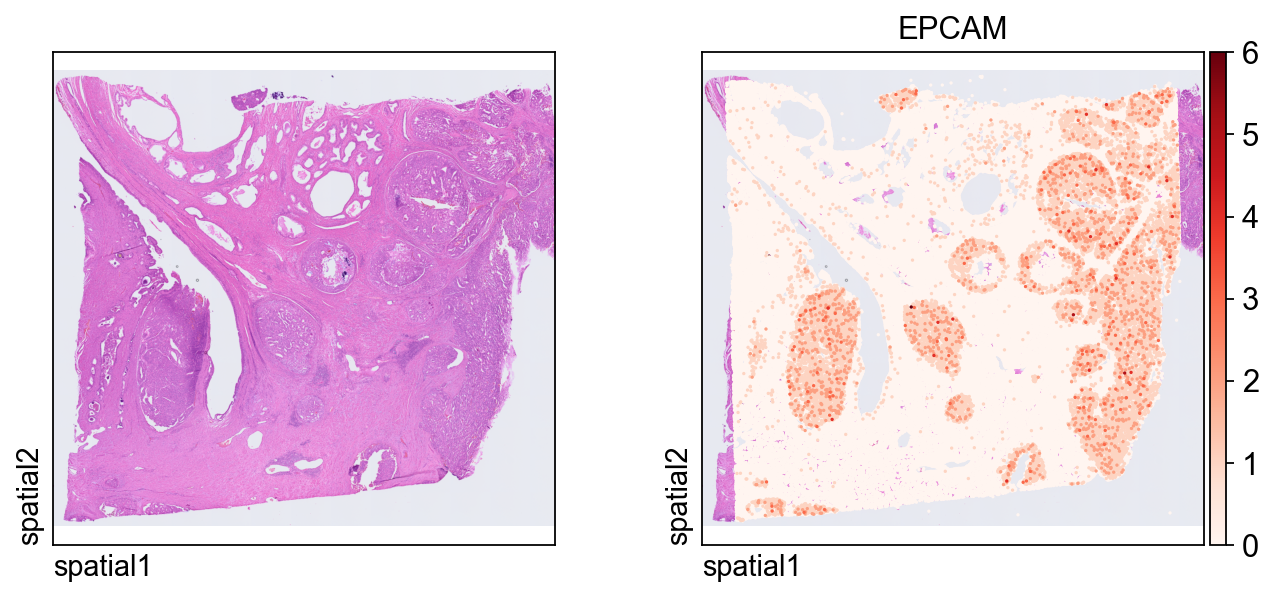
这张图在 segmentation-level 对象上展示同一个 marker EPCAM。由于每个观测现在对应一个分割细胞,因此空间模式会更容易从“细胞中心”的角度来解释。
ov.pl.spatial(
adata_seg, color=[None,"EPCAM"],
size=6, linewidth=0,
legend_fontsize=13, frameon=None,
cmap='Reds'
)
聚焦局部感兴趣区域#
全切片视图适合看整体背景,但很多时候,局部窗口更方便做细节检查。下面的辅助函数会根据空间坐标对子集进行筛选。
参数说明:
xlim和ylim:定义保留区域的矩形范围;adata.obsm["spatial"]:存储用于空间筛选的 x/y 坐标;obs_names_make_unique()和var_names_make_unique():避免子集化后出现重复标识符。
对于高密度的 Visium HD 数据来说,这种局部放大并不只是为了好看,而是判断图像结构、分割几何和基因表达是否一致的最快方式之一。
def subset_data(adata, xlim=(24500, 26000), ylim=(5000, 6000)):
x, y = adata.obsm["spatial"].T
bdata = adata[(xlim[0] <= x) & (x <= xlim[1]) & (ylim[0] <= y) & (y <= ylim[1])].copy()
bdata.obs_names_make_unique()
bdata.var_names_make_unique()
return bdata
bdata = subset_data(adata_seg)
接下来的几个面板会比较同一块局部分割区域的不同渲染方式。虽然这一步主要属于可视化调整,但它很适合帮助你判断最终图里应该保留多少分割边界信息。
这里的分割 polygon 没有显示可见边框(edges_width=0)。这种设置更强调表达信号本身,在视野很拥挤时通常是一个不错的默认方案。
ov.pl.spatialseg(
bdata,
color="EPCAM",
edges_color='white',
edges_width=0,
figsize=(6, 4),
alpha_img=1,
alpha=1,
legend_fontsize=13,
#cmap='Reds',
#img_key=False,
#alpha=1,
)
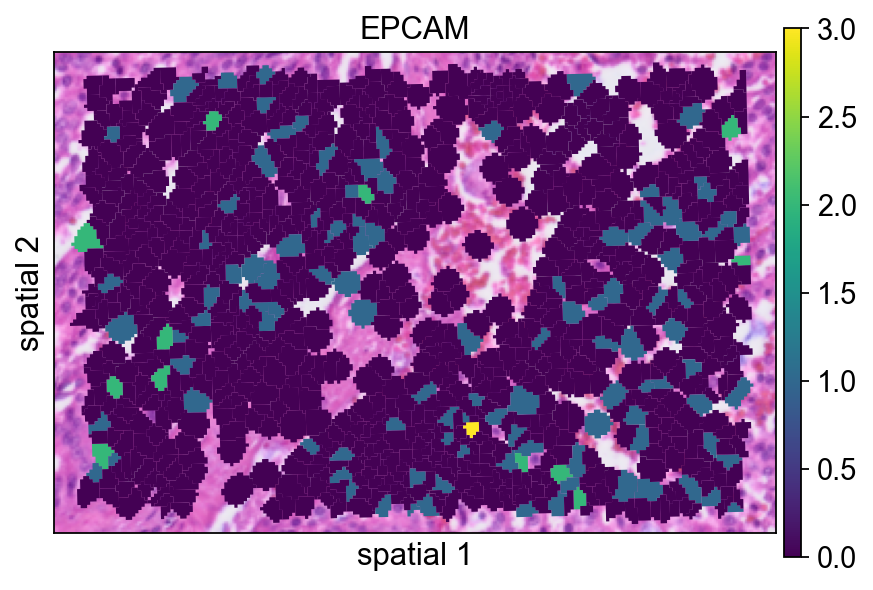
<Axes: title={'center': 'EPCAM'}, xlabel='spatial 1', ylabel='spatial 2'>
给每个 polygon 增加一条很细的边界(edges_width=0.1)有助于区分相邻细胞。当相邻 segment 的表达值比较接近时,这通常会让图更容易读。
ov.pl.spatialseg(
bdata,
color="EPCAM",
edges_color='white',
edges_width=0.1,
figsize=(6, 4),
alpha_img=1,
alpha=1,
legend_fontsize=13,
#cmap='Reds',
#img_key=False,
)
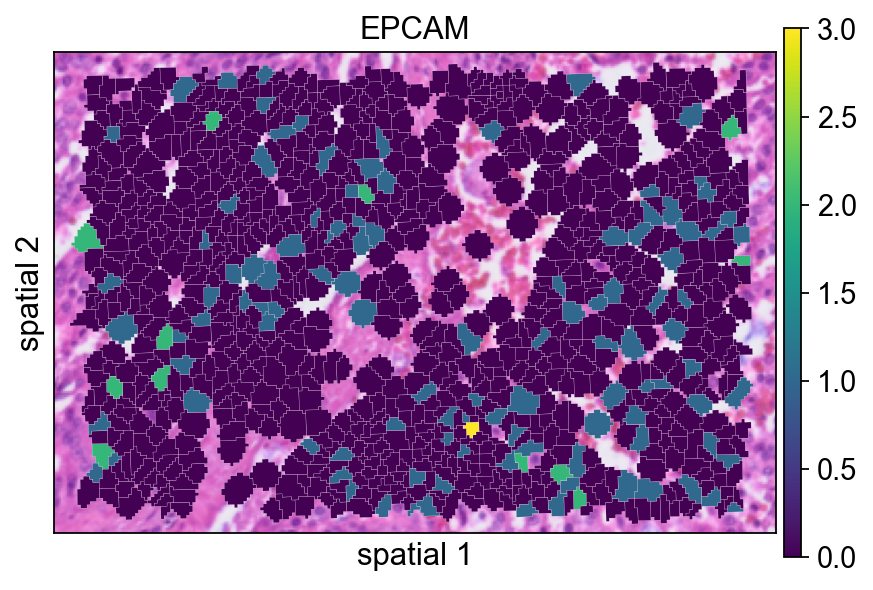
<Axes: title={'center': 'EPCAM'}, xlabel='spatial 1', ylabel='spatial 2'>
seg_contourpx 用像素单位控制分割轮廓的粗细。适当增大这个值能让边界更明显,尤其适合汇报图或背景组织纹理较复杂的情况。
ov.pl.spatialseg(
bdata,
color="EPCAM",
edges_color='white',
edges_width=0,
figsize=(6, 4),
#library_id='1',
alpha_img=1,
seg_contourpx=1.5,
alpha=1,
legend_fontsize=13,
)
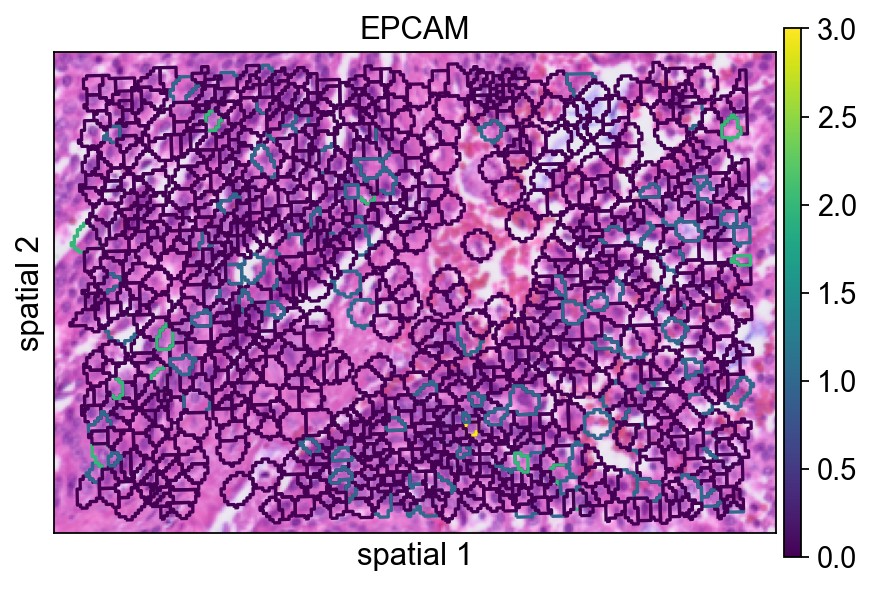
<Axes: title={'center': 'EPCAM'}, xlabel='spatial 1', ylabel='spatial 2'>
识别局部区域的空间变异基因#
接下来,我们在子集区域上运行 ov.space.svg()。
几个重要参数:
mode='moranI':使用 Moran’s I 按空间自相关对基因排序;n_svgs=3000:保留前 3000 个候选空间变异基因;n_perms=100:通过置换估计统计显著性;n_jobs=8:在 CPU worker 间并行计算。
bdata=ov.space.svg(
bdata,mode='moranI',
n_svgs=3000,
n_perms=100,n_jobs=8,
)
bdata
Spatial neighbors: 961 cells, 5582 connections (avg 5.8 neighbors/cell).
Stored in adata.obsp['spatial_connectivities'] and adata.obsp['spatial_distances'].
Stored 18132 gene results in adata.uns['moranI'].
AnnData object with n_obs × n_vars = 961 × 18132
obs: 'geometry'
var: 'gene_ids', 'feature_types', 'genome', 'moranI', 'moranI_pval', 'pval_adj', 'space_variable_features', 'highly_variable'
uns: 'spatial', 'omicverse_io', 'spatial_neighbors', 'REFERENCE_MANU', 'moranI'
obsm: 'spatial'
obsp: 'spatial_connectivities', 'spatial_distances'
下表可以快速查看排名靠前的基因。moranI 表示空间自相关强度,moranI_pval 记录显著性估计,space_variable_features 标记该基因是否被保留为空间特征。
bdata.var[['moranI','moranI_pval','space_variable_features']].sort_values('moranI',ascending=False)
| moranI | moranI_pval | space_variable_features | |
|---|---|---|---|
| CHGA | 0.422765 | 9.294550e-111 | True |
| NPY | 0.412106 | 2.276425e-105 | True |
| PLA2G2A | 0.387953 | 1.158365e-93 | True |
| COL1A1 | 0.343667 | 5.067257e-74 | True |
| KLK3 | 0.341580 | 3.733330e-73 | True |
| ... | ... | ... | ... |
| SNU13 | -0.033147 | 9.546428e-01 | False |
| TRIOBP | -0.034662 | 9.617609e-01 | False |
| SETD5 | -0.036784 | 9.701712e-01 | False |
| HNRNPL | -0.041451 | 9.833822e-01 | False |
| TXNIP | -0.043999 | 9.881957e-01 | False |
18132 rows × 3 columns
这里以 NPY 为例展示一个具有局部空间结构的基因。使用 segmentation 渲染后,更容易判断它的信号究竟是在相邻细胞群中连续分布,还是只表现为零散点位。
ov.pl.spatialseg(
bdata,
color="NPY",
edges_color='white',
edges_width=0.1,
figsize=(6, 4),
alpha_img=0.8,
alpha=1,
legend_fontsize=13,
)
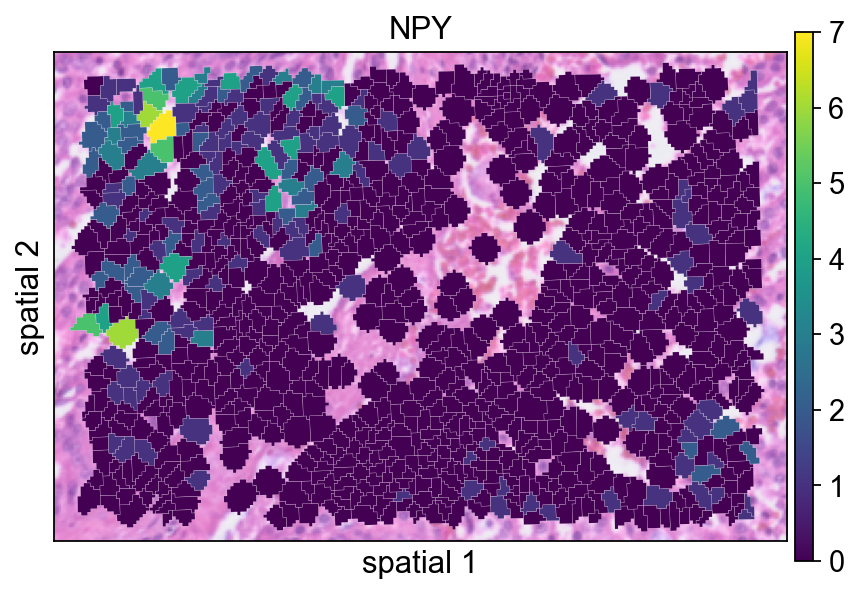
<Axes: title={'center': 'NPY'}, xlabel='spatial 1', ylabel='spatial 2'>
在归一化之前查看 X 的最大值,是一个简单但有用的 sanity check。它能帮助你快速感知当前子集对象中原始计数的大致量级。
bdata.X.max()
21.0
对局部子集做归一化和对数变换#
这里的标准预处理步骤是:
ov.pp.normalize_total(bdata):把每个观测归一化到可比较的文库大小;ov.pp.log1p(bdata):做 log(1+x) 变换,压缩动态范围。
这样处理后,空间表达图通常更容易在不同细胞之间进行比较。
ov.pp.normalize_total(bdata)
ov.pp.log1p(bdata)
🔍 Count Normalization:
Target sum: median
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 961 cells × 18,132 genes
✓ Runtime: 0.00s
完成归一化和 log 变换后,数值范围应明显小于原始矩阵,这说明变换已经生效。
bdata.X.max()
4.4248466
在预处理之后重新绘制 NPY,有助于判断归一化如何改变信号的视觉对比度。
ov.pl.spatialseg(
bdata,
color="NPY",
edges_color='white',
edges_width=0.1,
figsize=(6, 4),
alpha_img=0.8,
alpha=1,
legend_fontsize=13,
)
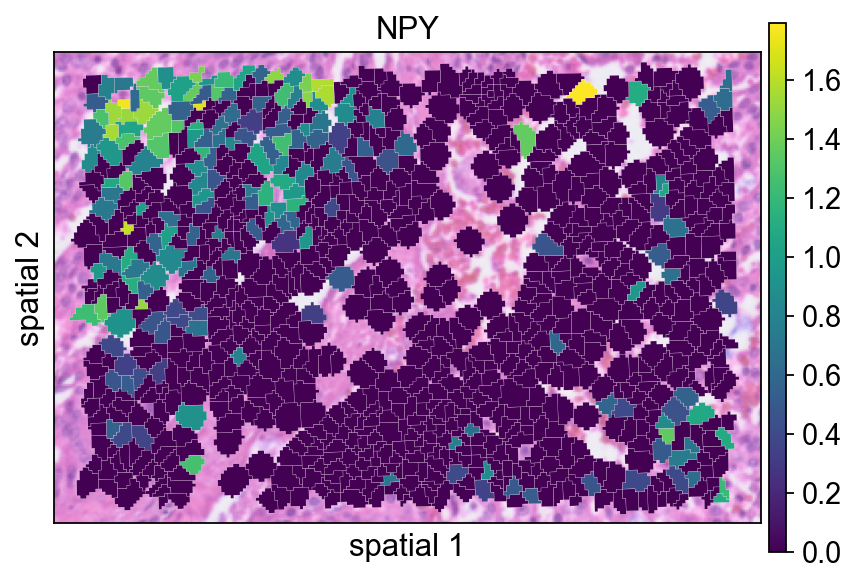
<Axes: title={'center': 'NPY'}, xlabel='spatial 1', ylabel='spatial 2'>
同一个基因也可以用 ov.pl.spatial() 展示,此时观测值会被渲染为点,而不是填充的分割 polygon。
这里有两个参数尤其有用:
size=1.5:控制每个观测的点大小;vmax='p99.2':把颜色上限裁剪到 99.2 百分位,减少极端值对视觉动态范围的影响。
fig, ax = ov.plt.subplots(figsize=(6, 4))
ov.pl.spatial(
bdata, color="NPY",
size=1.5, linewidth=0,
legend_fontsize=13, frameon=True,
cmap='Reds',vmax='p99.2',
ax=ax,
)
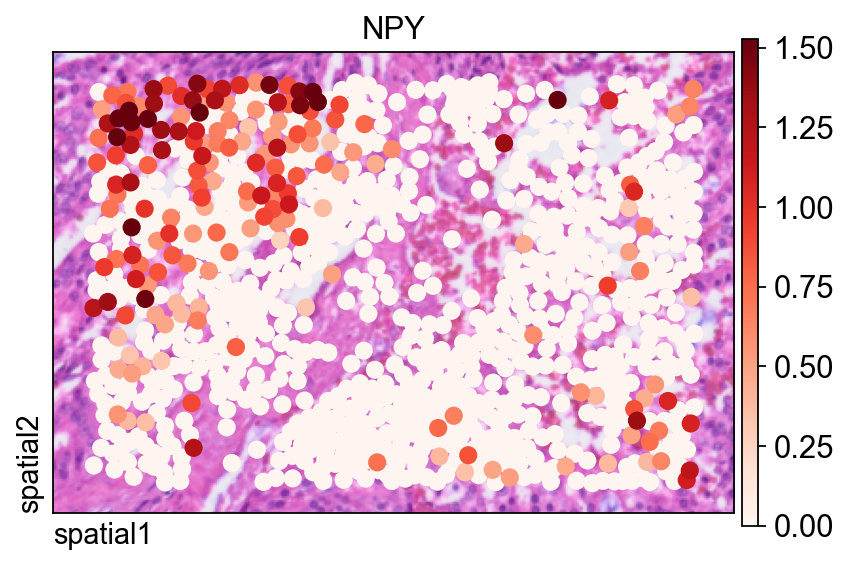
在完整 segmentation-level 数据集上计算空间变异基因#
在局部区域探索之后,我们对完整 segmentation 对象重复进行 SVG 检测。这里 notebook 使用 mode='pearsonr',它与 Moran’s I 不同,是另一种识别空间结构的标准。
adata=ov.space.svg(
adata_seg,mode='pearsonr',
n_svgs=3000,
)
adata
🔍 [2026-03-08 04:48:27] Running preprocessing in 'cpu-gpu-mixed' mode...
Begin robust gene identification
After filtration, 18109/18132 genes are kept.
Among 18109 genes, 13307 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 1,410 highly-expressed genes from normalization computation
⚠️ Warning: 620 cells have zero counts
✅ Count Normalization Completed Successfully!
✓ Processed: 227,257 cells × 13,307 genes
✓ Runtime: 7.77s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 3,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 3,000 highly variable genes out of 13,307 total (22.5%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 17.67 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 18.605s │
│ Shape: 227,257 x 18,132 -> 227,257 x 13,307 │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ REFERENCE_MANU │
│ │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │ ✚ status │
│ │ ✚ status_args │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 227257x13307) │
│ │
╰────────────────────────────────────────────────────────────────────╯
AnnData object with n_obs × n_vars = 227257 × 18132
obs: 'geometry'
var: 'gene_ids', 'feature_types', 'genome', 'space_variable_features', 'highly_variable'
uns: 'spatial', 'omicverse_io', 'REFERENCE_MANU'
obsm: 'spatial'
同样地,在对完整数据集做预处理之前,先检查一下当前数值范围会更稳妥。
adata.X.max()
332.0
完整数据集会以与局部子集相同的方式完成归一化和 log 变换,这样后续降维和聚类就在可比较的尺度上进行。
ov.pp.normalize_total(adata)
ov.pp.log1p(adata)
🔍 Count Normalization:
Target sum: median
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 227,257 cells × 18,132 genes
✓ Runtime: 0.10s
将矩阵限制在空间变异基因上#
通过 adata.raw = adata 可以保留过滤前的完整对象,方便后续参考。下一步再把矩阵限制到 adata.var.space_variable_features 标记出的基因上,从而降低噪声,并让下游分析更聚焦于空间信息更强的特征。
%%time
adata.raw = adata
adata = adata[:, adata.var.space_variable_features]
adata
CPU times: user 13 ms, sys: 70.1 ms, total: 83.1 ms
Wall time: 82.1 ms
View of AnnData object with n_obs × n_vars = 227257 × 3000
obs: 'geometry'
var: 'gene_ids', 'feature_types', 'genome', 'space_variable_features', 'highly_variable'
uns: 'spatial', 'omicverse_io', 'REFERENCE_MANU', 'log1p'
obsm: 'spatial'
构建邻接图和 UMAP 嵌入#
这一单元遵循常见的流程:缩放、PCA、构图,再计算 UMAP 嵌入。
参数说明:
PCA 中的
n_pcs=50:保留前 50 个主成分;n_neighbors=15:定义构图时局部邻域的大小;use_rep='scaled|original|X_pca':指定ov.pp.neighbors()在这一工作流约定下应使用哪种处理后的表示。实际执行时,这里依赖的是上一步刚生成的 PCA 表示。
%%time
ov.pp.scale(adata)
ov.pp.pca(adata,layer='scaled',n_pcs=50)
ov.pp.neighbors(adata, n_neighbors=15, n_pcs=50,
use_rep='scaled|original|X_pca')
ov.pp.umap(adata)
Converting scaled data to csr_matrix format...
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 29.9081s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ status │
│ │ ✚ status_args │
│ │
│ ● LAYERS │ ✚ scaled (sparse matrix, 227257x3000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🚀 Using GPU to calculate PCA...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA A100-SXM4-40GB
------------------------------ 4/40960 MiB (0.0%)
computing PCA🔍
with n_comps=50
Using CUDA device: NVIDIA A100-SXM4-40GB
✅ Using built-in torch_pca for GPU-accelerated PCA
🚀 Using torch_pca PCA for CUDA GPU acceleration
🚀 torch_pca PCA backend: CUDA GPU acceleration (supports sparse matrices)
📊 PCA input data type: SparseCSRView, shape: (227257, 3000), dtype: float64
📊 Sparse matrix density: 100.00%
finished✅ (2227.14s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 2227.5208s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ pca │
│ │ └─ params: {'zero_center': True, 'use_highly_variable': Tr...│
│ │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ X_pca (array, 227257x50) │
│ │ ✚ scaled|original|X_pca (array, 227257x50) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🚀 Using torch CPU/GPU mixed mode to calculate neighbors...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA A100-SXM4-40GB
------------------------------ 425/40960 MiB (1.0%)
🔍 K-Nearest Neighbors Graph Construction:
Mode: cpu-gpu-mixed
Neighbors: 15
Method: torch
Metric: euclidean
Representation: scaled|original|X_pca
PCs used: 50
🔍 Computing neighbor distances...
🔍 Computing connectivity matrix...
💡 Using UMAP-style connectivity
✓ Graph is fully connected
✅ KNN Graph Construction Completed Successfully!
✓ Processed: 227,257 cells with 15 neighbors each
✓ Results added to AnnData object:
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
╭─ SUMMARY: neighbors ───────────────────────────────────────────────╮
│ Duration: 52.9185s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ neighbors │
│ │ └─ params: {'n_neighbors': 15, 'method': 'torch', 'random_...│
│ │
│ ● OBSP │ ✚ connectivities (sparse matrix, 227257x227257) │
│ │ ✚ distances (sparse matrix, 227257x227257) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🔍 [2026-03-08 05:27:26] Running UMAP in 'cpu-gpu-mixed' mode...
🚀 Using torch GPU to calculate UMAP...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA A100-SXM4-40GB
------------------------------ 425/40960 MiB (1.0%)
🔍 UMAP Dimensionality Reduction:
Mode: cpu-gpu-mixed
Method: pumap
Components: 2
Min distance: 0.5
{'n_neighbors': 15, 'method': 'torch', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'scaled|original|X_pca', 'n_pcs': 50}
⚠️ Connectivities matrix was not computed with UMAP method
🔍 Computing UMAP parameters...
🔍 Computing UMAP embedding (Parametric PyTorch method)...
Using device: cuda
Dataset: 227257 samples × 50 features
Batch size: 512
Learning rate: 0.001
Training parametric UMAP model...
============================================================
🚀 Parametric UMAP Training
============================================================
📊 Device: cuda
📈 Data shape: torch.Size([227257, 50])
🔗 Building UMAP graph...
🚀 Using PyTorch Geometric KNN (faster)
🏋️ Starting Training...
────────────────────────────────────────────────────────────
✓ New best loss: 0.2845
✓ New best loss: 0.2564
✓ New best loss: 0.2523
✓ New best loss: 0.2500
✓ New best loss: 0.2483
✓ New best loss: 0.2468
✓ New best loss: 0.2455
✓ New best loss: 0.2445
✓ New best loss: 0.2434
✓ New best loss: 0.2425
✓ New best loss: 0.2416
✓ New best loss: 0.2409
✓ New best loss: 0.2402
✓ New best loss: 0.2396
✓ New best loss: 0.2389
✓ New best loss: 0.2386
✓ New best loss: 0.2380
✓ New best loss: 0.2375
✓ New best loss: 0.2369
✓ New best loss: 0.2366
────────────────────────────────────────────────────────────
✅ Training Completed!
📉 Final best loss: 0.2366
============================================================
💡 Using Parametric UMAP (PyTorch) on cuda
✅ UMAP Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 227,257 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_umap': UMAP coordinates (adata.obsm)
• 'umap': UMAP parameters (adata.uns)
✅ UMAP completed successfully.
╭─ SUMMARY: umap ────────────────────────────────────────────────────╮
│ Duration: 142.413s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ umap │
│ │ └─ params: {'a': 0.583030019901822, 'b': 1.3341669931033755}│
│ │
│ ● OBSM │ ✚ X_umap (array, 227257x2) │
│ │
╰────────────────────────────────────────────────────────────────────╯
⚙️ Using torch CPU/GPU mixed mode to calculate Leiden...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA A100-SXM4-40GB
------------------------------ 575/40960 MiB (1.4%)
Using batch size `n_batches` calculated from sqrt(n_obs): 476
Running GPU Leiden (batched)
Device: cpu
done: 54 clusters (0:04:36)
╭─ SUMMARY: leiden ──────────────────────────────────────────────────╮
│ Duration: 276.2389s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ leiden (category) │
│ │
│ ● UNS │ ✚ leiden │
│ │ └─ params: {'resolution': 1.0, 'random_state': 0, 'local_i...│
│ │
╰────────────────────────────────────────────────────────────────────╯
CPU times: user 1h 49min 38s, sys: 27min 27s, total: 2h 17min 5s
Wall time: 45min 29s
Leiden 聚类会把图结构划分成多个社区。resolution 参数控制聚类粒度:值越低,簇越少且更粗;值越高,划分越细。
ov.pp.leiden(adata,resolution=0.3)
⚙️ Using torch CPU/GPU mixed mode to calculate Leiden...
NVIDIA CUDA GPUs detected:
📊 [CUDA 0] NVIDIA A100-SXM4-40GB
------------------------------ 575/40960 MiB (1.4%)
Using batch size `n_batches` calculated from sqrt(n_obs): 476
Running GPU Leiden (batched)
Device: cpu
done: 13 clusters (0:05:32)
╭─ SUMMARY: leiden ──────────────────────────────────────────────────╮
│ Duration: 332.9071s │
│ Shape: 227,257 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
╰────────────────────────────────────────────────────────────────────╯
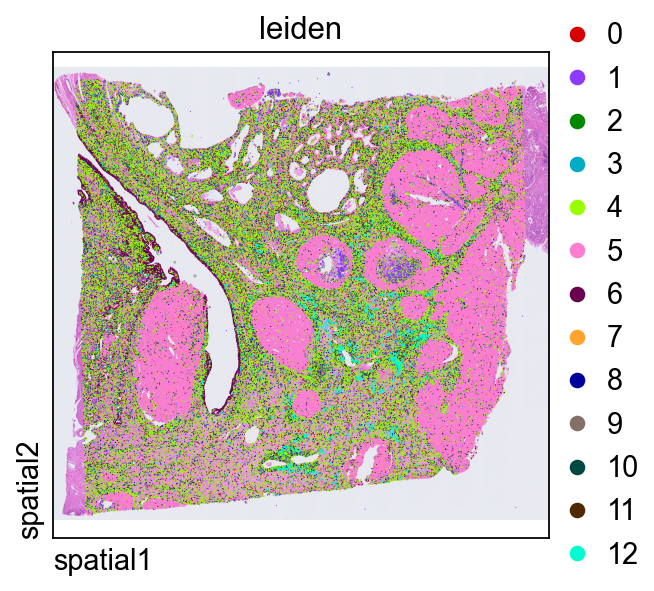
直接在组织空间中绘制 leiden,是判断转录组簇是否与连贯的解剖或组织学区域对应的快捷方式。
ov.pl.spatial(
adata, color=["leiden"],
size=2, linewidth=0,
legend_fontsize=13, frameon=None,
cmap='Reds'
)
在同一局部区域中放大查看聚类结果#
最后,我们把已经聚类的数据对象裁剪到前面相同的空间窗口中。这样更方便把聚类结果与原始 marker 表达和分割边界做直接比较。
def subset_data(adata, xlim=(24500, 26000), ylim=(5000, 6000)):
x, y = adata.obsm["spatial"].T
bdata = adata[(xlim[0] <= x) & (x <= xlim[1]) & (ylim[0] <= y) & (y <= ylim[1])].copy()
bdata.obs_names_make_unique()
bdata.var_names_make_unique()
return bdata
cdata = subset_data(adata)
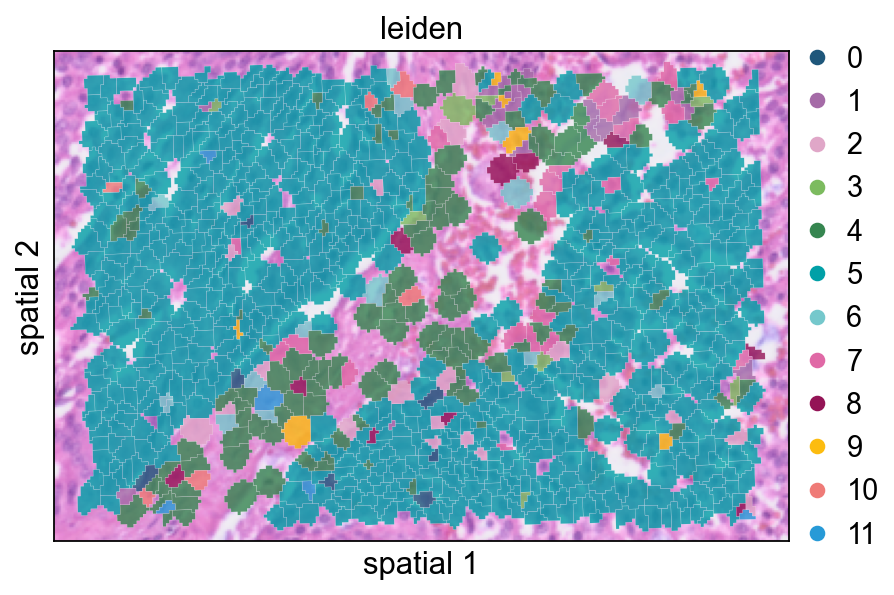
这个视图把聚类标签以部分透明的方式叠加在 segmentation polygon 上,适合检查簇与簇之间的过渡是否贴合可见的组织结构。
ov.pl.spatialseg(
cdata,
color="leiden",
edges_color='white',
edges_width=0.1,
figsize=(6, 4),
alpha_img=0.8,
alpha=0.8,
legend_fontsize=13,
palette=ov.pl.sc_color
)
<Axes: title={'center': 'leiden'}, xlabel='spatial 1', ylabel='spatial 2'>
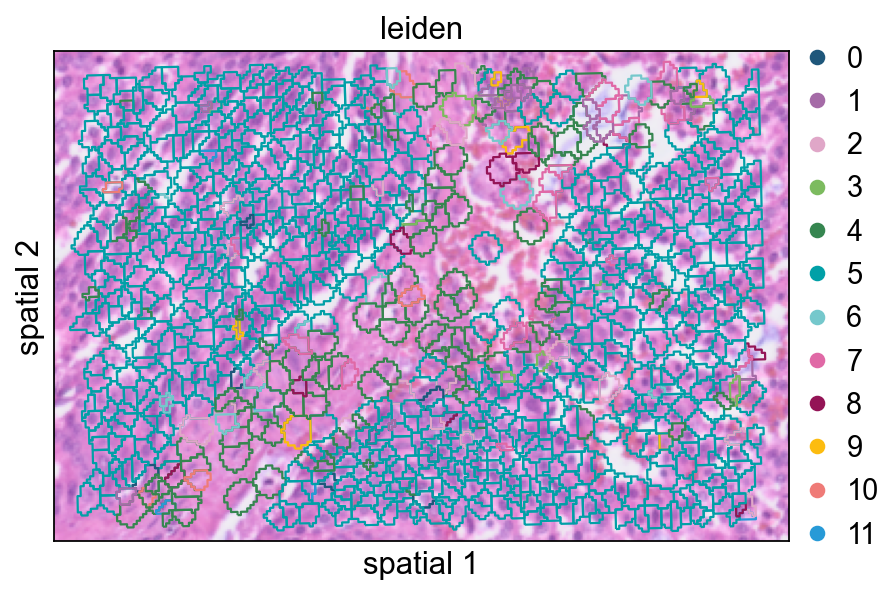
增大 seg_contourpx 会让 polygon 边界更锐利,这有助于观察相邻簇之间更细的分界。
ov.pl.spatialseg(
cdata,
color="leiden",
edges_color='white',
edges_width=0.1,
figsize=(6, 4),
alpha_img=0.8,
alpha=1,
legend_fontsize=13,
palette=ov.pl.sc_color,
seg_contourpx=1,
)
<Axes: title={'center': 'leiden'}, xlabel='spatial 1', ylabel='spatial 2'>
总结#
在这个 notebook 中,我们:
同时导入了 bin-level 和 segmentation-level 的 Visium HD 数据;
比较了不同的空间可视化方式;
在局部和全局两个尺度上识别了空间变异基因;
对数据完成归一化,并把分析限制到空间特征上;
构建了低维表示,并对分割细胞进行聚类。
综合来看,这套流程为 OmicVerse 中的 Visium HD 分析提供了一个很实用的起点:先做图像感知的质量检查,再做空间特征筛选,最后建立细胞层面的低维表示以支撑后续解释。