批量 RNA-seq 到单细胞 RNA-seqBulk2Single 用于批量 RNA-seq 反卷积。我们从 Bulk2Space 算法中提取了 beta-VAE 部分,并构建了一种可以从批量 RNA-seq 反卷积到单细胞 RNA-seq 的算法。此外,我们重新设计了数据的输入和输出,使其更兼容 Python 环境中的分析约定。Paper: De novo analysis of bulk RNA-seq data at spatially resolved single-cell resolutionCode: https://github.com/ZJUFanLab/bulk2spaceColab_Reproducibility:https://colab.research.google.com/drive/1He71hAyeAv1DHQyXUlxtoJ4QvwZwW7I0?usp=sharing本教程介绍了如何从批量 RNA-seq 和参考 scRNA-seq 数据中读取、设置和训练模型。我们使用 pdac 数据集作为示例#
import scanpy as scimport omicverse as ovimport matplotlib.pyplot as pltov.plot_set()
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
Version: 1.6.3, Tutorials: https://omicverse.readthedocs.io/
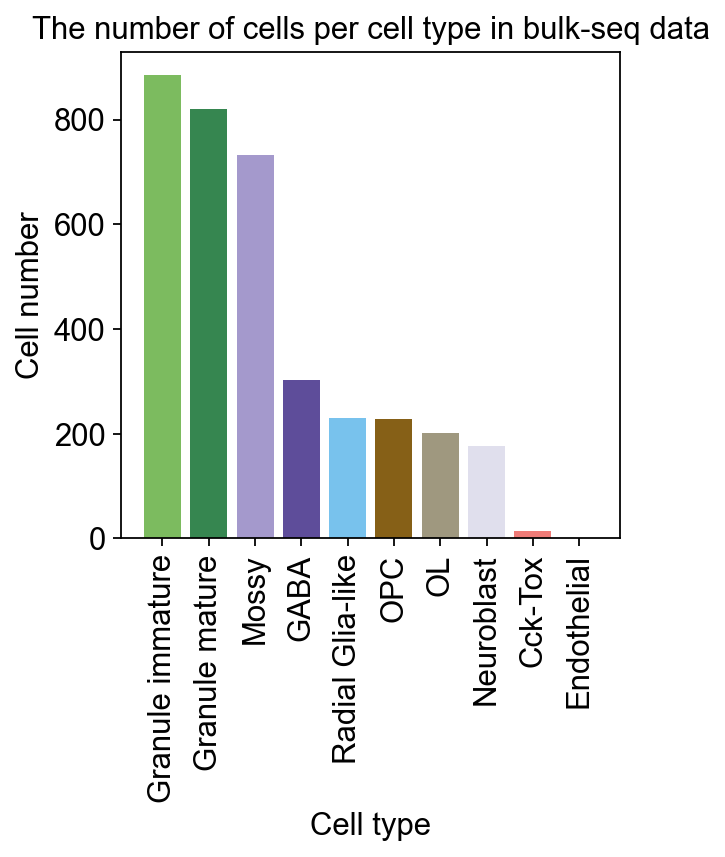
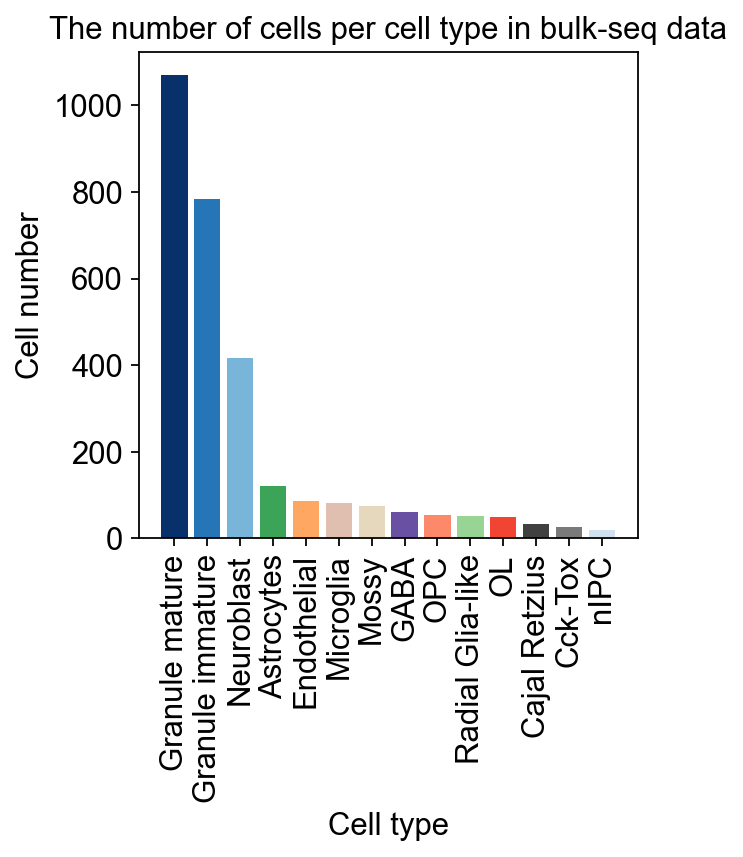
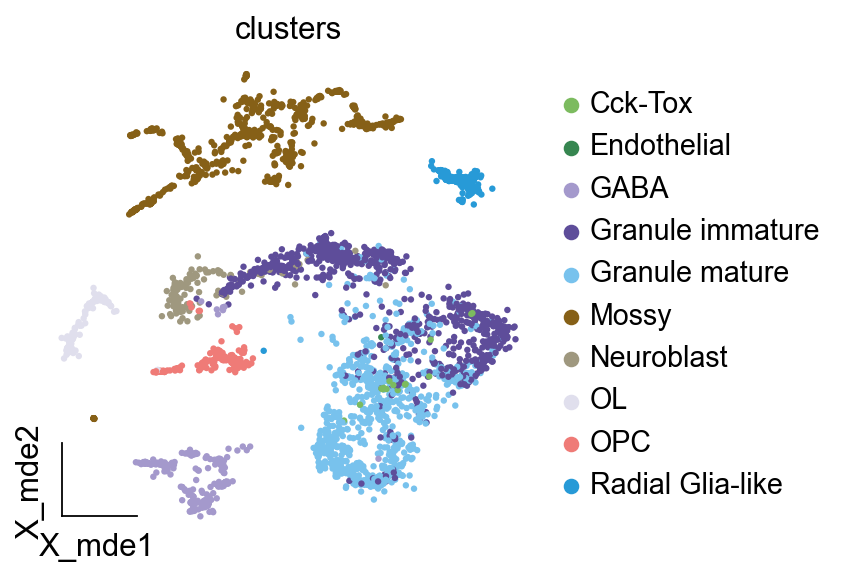
加载数据为了说明,我们对牙状回神经发生进行差异动力学分析,其包含多个异质亚群。我们利用从大鼠海马牙状回获得的单细胞 RNA-seq 数据 (GEO 编号: GSE95753) 以及批量 RNA-seq 数据 (GEO 编号: GSE74985)。#
bulk_data=ov.read('data/GSE74985_mergedCount.txt.gz',index_col=0)bulk_data=ov.bulk.Matrix_ID_mapping(bulk_data,'genesets/pair_GRCm39.tsv')bulk_data.head()
| dg_d_1 | dg_d_2 | dg_d_3 | dg_v_1 | dg_v_2 | dg_v_3 | ca4_1 | ca4_2 | ca4_3 | ca3_d_1 | ... | ca3_v_3 | ca2_1 | ca2_2 | ca2_3 | ca1_d_1 | ca1_d_2 | ca1_d_3 | ca1_v_1 | ca1_v_2 | ca1_v_3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gm12150 | 0 | 2 | 0 | 11 | 0 | 9 | 72 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mir219a-2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hspd1 | 1418 | 685 | 1404 | 3073 | 2316 | 1945 | 7724 | 8255 | 6802 | 4956 | ... | 8154 | 7104 | 5854 | 7508 | 5322 | 6172 | 5199 | 1865 | 1253 | 2298 |
| Crhbp | 0 | 0 | 0 | 31 | 17 | 32 | 0 | 0 | 0 | 29 | ... | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gm11735 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ... | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
5 rows × 24 columns
import anndataimport scvelo as scvsingle_data=scv.datasets.dentategyrus()single_data
AnnData object with n_obs × n_vars = 2930 × 13913
obs: 'clusters', 'age(days)', 'clusters_enlarged'
uns: 'clusters_colors'
obsm: 'X_umap'
layers: 'ambiguous', 'spliced', 'unspliced'
细胞比例计算我们现在可以设置 Bulk2Single 对象,这将确保模型所需的一切都准备好进行训练。我们需要指定 scRNA-seq 的细胞类型以反卷积批量 RNA-seq。并为每个细胞类型指定用于训练的标记基因数量。如果设置 gpu=-1,它将使用 CPU 配置 VAE 模型。#
model=ov.bulk2single.Bulk2Single(bulk_data=bulk_data,single_data=single_data, celltype_key='clusters',bulk_group=['dg_d_1','dg_d_2','dg_d_3'], top_marker_num=200,ratio_num=1,gpu=0)
在这里,我们改进了 Bulk2space 中细胞比例的估计,并消除了原始作者使用的回归估计,这通常会导致比例出现较大偏差,正如我们的分析所证实的。我们引入了 TAPE,该模型能够准确地将批量 RNA-seq 数据反卷积为细胞分数,并基于 scRNA-seq 数据在细胞类型水平预测细胞类型特异性基因表达。Paper: Deep autoencoder for interpretable tissue-adaptive deconvolution and cell-type-specific gene analysisCode: poseidonchan/TAPE
CellFractionPrediction=model.predicted_fraction()
Reading single-cell dataset, this may take 1 min
Reading dataset is done
Normalizing raw single cell data with scanpy.pp.normalize_total
Generating cell fractions using Dirichlet distribution without prior info (actually random)
RANDOM cell fractions is generated
You set sparse as True, some cell's fraction will be zero, the probability is 0.5
Sampling cells to compose pseudo-bulk data
Sampling is done
Reading training data
Reading is done
Reading test data
Reading test data is done
Using counts data to train model
Cutting variance...
Finding intersected genes...
Intersected gene number is 12227
Scaling...
Using minmax scaler...
training data shape is (5000, 12227)
test data shape is (24, 12227)
train model256 now
train model512 now
train model1024 now
Training of Scaden is done
Predicted Total Cell Num: 2457.268449380651
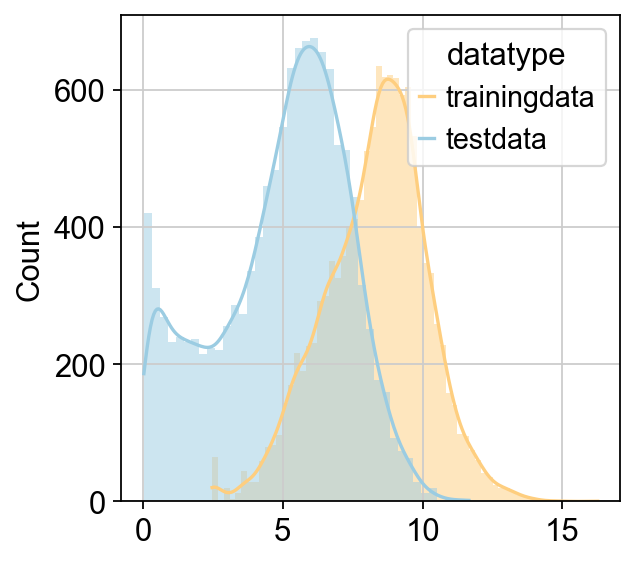
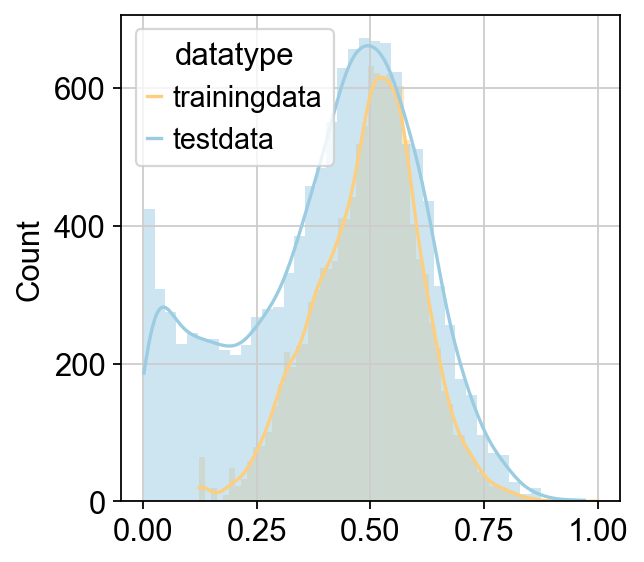
CellFractionPrediction.head()
| Astrocytes | Cajal Retzius | Cck-Tox | Endothelial | GABA | Granule immature | Granule mature | Microglia | Mossy | Neuroblast | OL | OPC | Radial Glia-like | nIPC | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dg_d_1 | 0.004780 | 0.003839 | 0.004187 | 0.002460 | 0.005536 | 0.527208 | 0.393742 | 0.005203 | 0.028935 | 0.004639 | 0.007397 | 0.005216 | 0.002961 | 0.003898 |
| dg_d_2 | 0.005013 | 0.002877 | 0.003001 | 0.002407 | 0.004481 | 0.508747 | 0.413222 | 0.004478 | 0.032327 | 0.006355 | 0.007488 | 0.004283 | 0.002102 | 0.003218 |
| dg_d_3 | 0.003915 | 0.002676 | 0.002945 | 0.002558 | 0.005772 | 0.479360 | 0.446842 | 0.004949 | 0.026702 | 0.006624 | 0.008542 | 0.004052 | 0.002157 | 0.002908 |
| dg_v_1 | 0.003247 | 0.002842 | 0.003309 | 0.001613 | 0.010134 | 0.539566 | 0.347792 | 0.002481 | 0.063813 | 0.006122 | 0.008335 | 0.005785 | 0.002116 | 0.002846 |
| dg_v_2 | 0.004015 | 0.003188 | 0.003747 | 0.002137 | 0.010382 | 0.523644 | 0.362331 | 0.002693 | 0.056484 | 0.009367 | 0.008487 | 0.007403 | 0.002478 | 0.003644 |
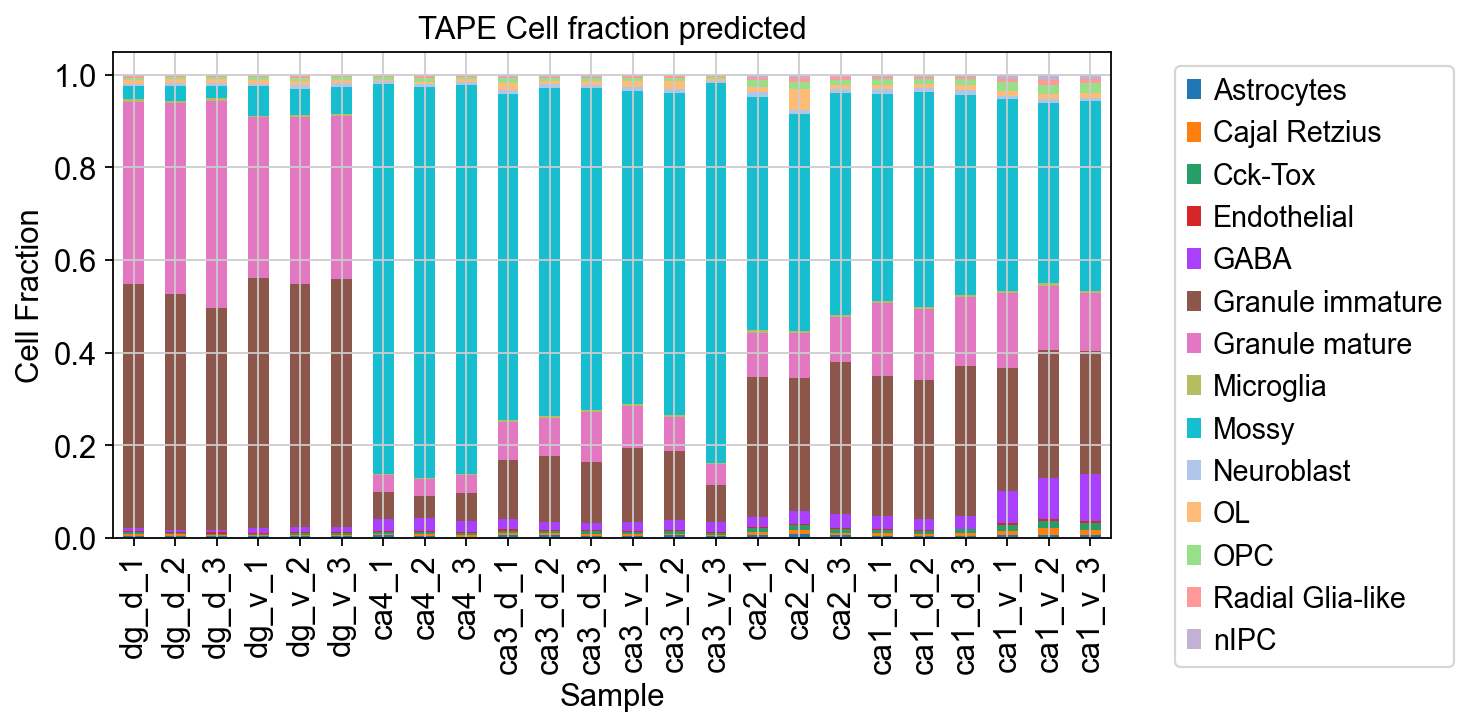
我们使用堆积直方图来可视化每个样品的细胞比例
ax = CellFractionPrediction.plot(kind='bar', stacked=True, figsize=(8, 4))ax.set_xlabel('Sample')ax.set_ylabel('Cell Fraction')ax.set_title('TAPE Cell fraction predicted')plt.legend(bbox_to_anchor=(1.05, 1),ncol=1,)plt.show()
Bulk2single 训练### 预处理单细胞 RNA-seq 和批量 RNA-seq获得每个样品的细胞比例后,我们还想获得样品的单细胞数据,其中我们使用 beta-VAE 来预测批量数据中的细胞,我们首先对数据进行了预处理。这些组是 [‘dg_d_1’, ‘dg_d_2’, ‘dg_d_3’],代表样品 DG 颗粒细胞#
model.bulk_preprocess_lazy()model.single_preprocess_lazy()model.prepare_input()
......drop duplicates index in bulk data
......deseq2 normalize the bulk data
......log10 the bulk data
......calculate the mean of each group
......normalize the single data
normalizing counts per cell
finished (0:00:00)
......log1p the single data
......prepare the input of bulk2single
...loading data
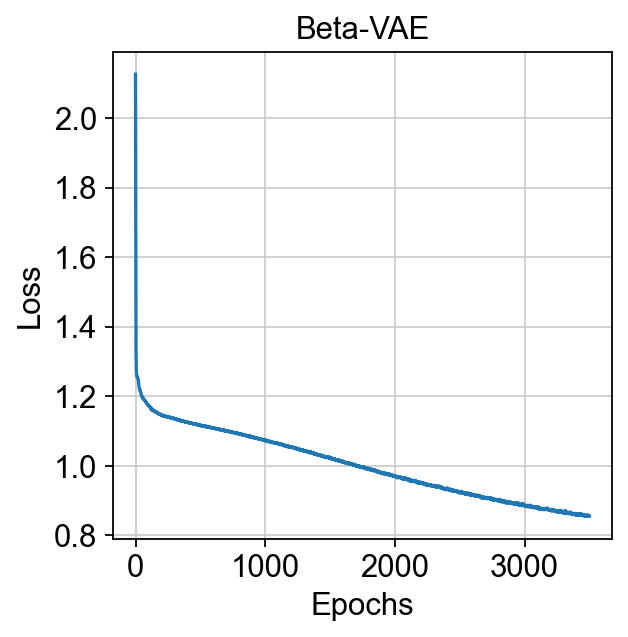
训练 VAE 模型我们开始训练 VAE 模型以生成单细胞数据,该过程在 CPU 上花费约 3 小时,在 GPU 上仅需 10 分钟。
Note
默认最大周期设置为 3500,但实际上 Bulk2Single 一旦模型收敛就会提前停止,这很少需要那么多,特别是对于大型数据集。(我们可以设置 patience 来控制停止步骤)