使用 SCSA 进行细胞类型自动注释#
单细胞转录组技术可以在一次实验中分析成千上万个细胞,并识别多种组织和物种中的新型细胞类型、细胞状态及其动态变化。围绕组织单细胞转录组图谱的构建,已经发展出较为标准的实验方案和分析流程。
本教程重点介绍如何解释这类数据,以识别细胞类型、细胞状态以及其他具有生物学意义的模式,从而构建带注释的细胞图谱。
论文: [SCSA: A Cell Type Annotation Tool for Single-Cell RNA-seq Data](https://doi.org/10.3389/fgene.2020.00490
)
Colab 复现: https://colab.research.google.com/drive/1BC6hPS0CyBhNu0BYk8evu57-ua1bAS0T?usp=sharing
注意
SCSA 不适用于稀有细胞类型注释。
import scanpy as sc
print(f'scanpy version:{sc.__version__}')
import omicverse as ov
print(f'omicverse version:{ov.__version__}')
ov.ov_plot_set()
scanpy version:1.11.5
omicverse version:2.1.2rc1
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.2rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
加载数据#
该数据集由来自一位健康供体的 3k PBMC 组成,可从 10x Genomics 免费获取(可通过这个下载链接或对应的数据页面下载)。在 Unix 系统中,你可以取消下面代码的注释并运行,以下载和解压数据。最后一行会创建一个用于保存处理后数据的目录。
# !mkdir data
# !wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
# !cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
# !mkdir write
将 count matrix 读入 AnnData 对象。AnnData 提供了多个槽位来存储注释信息和不同形式的数据表示,并且自带基于 HDF5 的文件格式:.h5ad。
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/', # the directory with the `.mtx` file
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True) # write a cache file for faster subsequent reading
数据预处理#
这里我们使用 ov.single.scanpy_lazy 对 scRNA-seq 原始数据进行预处理,流程包括过滤双细胞、按细胞归一化、log1p 转换、提取高变基因以及细胞聚类计算。
如果你希望逐步体验预处理流程,我们也提供了更详细的分步示例,请参考我们的预处理章节。
其中原始 counts 保存在 count layer 中,原始数据本体可通过 adata.raw.to_adata() 访问。
#adata=ov.single.scanpy_lazy(adata)
#quantity control
adata=ov.pp.qc(
adata,
tresh={'mito_perc': 0.05, 'nUMIs': 500, 'detected_genes': 250}
)
#normalize and high variable genes (HVGs) calculated
adata=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
#save the whole genes and filter the non-HVGs
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features]
#scale the adata.X
ov.pp.scale(adata)
#Dimensionality Reduction
ov.pp.pca(adata,layer='scaled',n_pcs=50)
#Neighbourhood graph construction
sc.pp.neighbors(
adata,
n_neighbors=15,
n_pcs=50,
use_rep='scaled|original|X_pca'
)
#clusters
sc.tl.leiden(adata)
#Dimensionality Reduction for visualization(X_mde=X_umap+GPU)
X_mde = ov.utils.mde(adata.obsm["scaled|original|X_pca"])
if hasattr(X_mde, "detach"):
X_mde = X_mde.detach().cpu().numpy()
elif hasattr(X_mde, "cpu") and hasattr(X_mde, "numpy"):
X_mde = X_mde.cpu().numpy()
adata.obsm["X_mde"] = X_mde
adata
🖥️ Using CPU mode for QC...
📊 Step 1: Calculating QC Metrics
✓ Gene Family Detection:
┌──────────────────────────────┬────────────────────┬────────────────────┐
│ Gene Family │ Genes Found │ Detection Method │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Mitochondrial │ 13 │ Auto (MT-) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Ribosomal │ 106 │ Auto (RPS/RPL) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Hemoglobin │ 13 │ Auto (regex) │
└──────────────────────────────┴────────────────────┴────────────────────┘
✓ QC Metrics Summary:
┌─────────────────────────┬────────────────────┬─────────────────────────┐
│ Metric │ Mean │ Range (Min - Max) │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ nUMIs │ 2367 │ 548 - 15844 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Detected Genes │ 847 │ 212 - 3422 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Mitochondrial % │ 2.2% │ 0.0% - 22.6% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Ribosomal % │ 34.9% │ 1.1% - 59.4% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Hemoglobin % │ 0.0% │ 0.0% - 1.4% │
└─────────────────────────┴────────────────────┴─────────────────────────┘
📈 Original cell count: 2,700
🔧 Step 2: Quality Filtering (SEURAT)
Thresholds: mito≤0.05, nUMIs≥500, genes≥250
📊 Seurat Filter Results:
• nUMIs filter (≥500): 0 cells failed (0.0%)
• Genes filter (≥250): 3 cells failed (0.1%)
• Mitochondrial filter (≤0.05): 57 cells failed (2.1%)
✓ Filters applied successfully
✓ Combined QC filters: 60 cells removed (2.2%)
🎯 Step 3: Final Filtering
Parameters: min_genes=200, min_cells=3
Ratios: max_genes_ratio=1, max_cells_ratio=1
✓ Final filtering: 0 cells, 19,041 genes removed
🔍 Step 4: Doublet Detection
⚠️ Note: 'scrublet' detection is too old and may not work properly
💡 Consider using 'doublets_method=sccomposite' for better results
🔍 Running scrublet doublet detection...
🔍 Running Scrublet Doublet Detection:
Mode: cpu
Computing doublet prediction using Scrublet algorithm
🔍 Filtering genes and cells...
🔍 Filtering genes...
Parameters: min_cells≥3
✓ Filtered: 0 genes removed
🔍 Filtering cells...
Parameters: min_genes≥3
✓ Filtered: 0 cells removed
🔍 Normalizing data and selecting highly variable genes...
🔍 Count Normalization:
Target sum: median
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 2,640 cells × 13,697 genes
✓ Runtime: 0.01s
🔍 Highly Variable Genes Selection:
Method: seurat
⚠️ Gene indices [7846] fell into a single bin: normalized dispersion set to 1
💡 Consider decreasing `n_bins` to avoid this effect
✅ HVG Selection Completed Successfully!
✓ Selected: 1,738 highly variable genes out of 13,697 total (12.7%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'dispersions': Float vector (adata.var)
• 'dispersions_norm': Float vector (adata.var)
🔍 Simulating synthetic doublets...
🔍 Normalizing observed and simulated data...
🔍 Count Normalization:
Target sum: 1000000.0
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 2,640 cells × 1,738 genes
✓ Runtime: 0.00s
🔍 Count Normalization:
Target sum: 1000000.0
Exclude highly expressed: False
✅ Count Normalization Completed Successfully!
✓ Processed: 5,280 cells × 1,738 genes
✓ Runtime: 0.01s
🔍 Embedding transcriptomes using PCA...
📊 Scrublet PCA input data type (CPU) - X_obs: ndarray, shape: (2640, 1738), dtype: float64
📊 Scrublet PCA input data type (CPU) - X_sim: ndarray, shape: (5280, 1738), dtype: float64
🔍 Calculating doublet scores...
🔍 Calling doublets with threshold detection...
📊 Automatic threshold: 0.326
📈 Detected doublet rate: 1.3%
🔍 Detectable doublet fraction: 34.0%
📊 Overall doublet rate comparison:
• Expected: 5.0%
• Estimated: 3.9%
✅ Scrublet Analysis Completed Successfully!
✓ Results added to AnnData object:
• 'doublet_score': Doublet scores (adata.obs)
• 'predicted_doublet': Boolean predictions (adata.obs)
• 'scrublet': Parameters and metadata (adata.uns)
✓ Scrublet completed: 35 doublets removed (1.3%)
╭─ SUMMARY: qc ──────────────────────────────────────────────────────╮
│ Duration: 0.812s │
│ Shape: 2,700 x 32,738 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ cell_complexity (float) │
│ │ ✚ detected_genes (int) │
│ │ ✚ hb_perc (float) │
│ │ ✚ mito_perc (float) │
│ │ ✚ nUMIs (float) │
│ │ ✚ passing_mt (bool) │
│ │ ✚ passing_nUMIs (bool) │
│ │ ✚ passing_ngenes (bool) │
│ │ ✚ ribo_perc (float) │
│ │
│ ● VAR │ ✚ hb (bool) │
│ │ ✚ mt (bool) │
│ │ ✚ ribo (bool) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🔍 [2026-04-08 19:00:21] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 13697/13697 genes are kept.
Among 13697 genes, 13696 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 0 highly-expressed genes from normalization computation
Excluded genes: []
✅ Count Normalization Completed Successfully!
✓ Processed: 2,605 cells × 13,696 genes
✓ Runtime: 0.04s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 2,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 2,000 highly variable genes out of 13,696 total (14.6%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 0.12 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 0.1281s │
│ Shape: 2,605 x 13,697 -> 2,605 x 13,696 │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 2605x13696) │
│ │
╰────────────────────────────────────────────────────────────────────╯
Converting scaled data to csr_matrix format...
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.0872s │
│ Shape: 2,605 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (sparse matrix, 2605x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=50
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: SparseCSRMatrixView, shape: (2605, 2000), dtype: float64
📊 Sparse matrix density: 100.00%
🔧 PCA solver used: covariance_eigh
finished✅ (16.44s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 16.4474s │
│ Shape: 2,605 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ pca │
│ │ └─ params: {'zero_center': True, 'use_highly_variable': Tr...│
│ │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ X_pca (array, 2605x50) │
│ │ ✚ scaled|original|X_pca (array, 2605x50) │
│ │
╰────────────────────────────────────────────────────────────────────╯
AnnData object with n_obs × n_vars = 2605 × 2000
obs: 'nUMIs', 'mito_perc', 'ribo_perc', 'hb_perc', 'detected_genes', 'cell_complexity', 'passing_mt', 'passing_nUMIs', 'passing_ngenes', 'doublet_score', 'predicted_doublet', 'leiden'
var: 'gene_ids', 'mt', 'ribo', 'hb', 'n_cells', 'percent_cells', 'robust', 'highly_variable_features', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable'
uns: 'scrublet', 'status', 'status_args', 'REFERENCE_MANU', 'history_log', 'log1p', 'hvg', 'pca', 'scaled|original|pca_var_ratios', 'scaled|original|cum_sum_eigenvalues', 'neighbors', 'leiden'
obsm: 'X_pca', 'scaled|original|X_pca', 'X_mde'
varm: 'PCs', 'scaled|original|pca_loadings'
layers: 'counts', 'scaled'
obsp: 'distances', 'connectivities'
自动细胞注释#
我们基于 adata 创建一个 pySCSA 对象,并设置若干参数以获得正确的注释结果。
在常规注释任务中,通常将 celltype 设为 'normal',并将 target 设为 'cellmarker' 或 'panglaodb' 来进行细胞类型注释。
而在肿瘤注释任务中,则需要将 celltype 设为 'cancer',并将 target 设为 'cancersea'。
说明
使用 SCSA 进行注释前需要先下载数据库。通常可以自动下载,但在某些网络环境下可能会遇到下载失败的问题。
2023 版本(基于 pandas<=1.5.3):数据库可从 figshare、Google Drive 和 百度云 下载。
2024 版本(基于 pandas>2):数据库可从 Google Drive 和 百度云 下载。
并且你需要将参数 model_path 设置为对应数据库文件路径。
数据库构建代码可参考 scsa_database_create.ipynb。感谢 @fredsamhaak 与 @H1207953831 在 issue #232 和 #176 中的讨论与帮助。
scsa=ov.single.pySCSA(
adata=adata,
foldchange=1.5,
pvalue=0.01,
celltype='normal',
target='cellmarker',
tissue='All',
model_path='temp/pySCSA_2024_v1_plus.db'
)
在前面的聚类步骤中,我们使用的是 leiden 算法,因此这里将聚类类型指定为 leiden。如果你使用的是 louvain,请相应修改该参数。另外,这里会对所有 cluster 进行注释;如果你只想注释部分 cluster,可按 '[1]'、'[1,2,3]'、'[...]' 这样的格式输入。
rank_rep 对应 sc.tl.rank_genes_groups(adata, clustertype, method='wilcoxon') 的计算过程;如果你的 adata.uns 中已经包含 rank_genes_groups 结果,则可将 rank_rep 设为 False。
anno=scsa.cell_anno(
clustertype='leiden',
cluster='all',
rank_rep=True
)
ranking genes
finished (0:00:00)
...Auto annotate cell
🔍 Version V2.2 [2024/12/18]
📊 DB load: GO_items:47347, Human_GO:3, Mouse_GO:3,
CellMarkers:82887, CancerSEA:1574, PanglaoDB:24223
Ensembl_HGNC:61541, Ensembl_Mouse:55414
<omicverse.single._SCSA.Annotator object at 0x13051f690>
🔍 Version V2.2 [2024/12/18]
📊 DB load: GO_items:47347, Human_GO:3, Mouse_GO:3,
CellMarkers:82887, CancerSEA:1574, PanglaoDB:24223
Ensembl_HGNC:61541, Ensembl_Mouse:55414
📦 Load markers: 70276
============================================================
🔬 Analyzing 9 clusters...
============================================================
[1/9] Cluster 0 │ 75 genes │ 1351 other genes
[2/9] Cluster 1 │ 154 genes │ 1292 other genes
[3/9] Cluster 2 │ 581 genes │ 1250 other genes
[4/9] Cluster 3 │ 128 genes │ 1307 other genes
[5/9] Cluster 4 │ 81 genes │ 1370 other genes
[6/9] Cluster 5 │ 908 genes │ 989 other genes
[7/9] Cluster 6 │ 256 genes │ 1265 other genes
[8/9] Cluster 7 │ 52 genes │ 1384 other genes
[9/9] Cluster 8 │ 5 genes │ 1384 other genes
============================================================
✅ Cluster analysis completed! (9/9 processed)
============================================================
================================================================================
📋 Cell Type Annotation Results
================================================================================
Cluster Type Cell Type Score Times
--------------------------------------------------------------------------------
0 ⚠️ ? T cell|Naive CD8+ T cell 8.781928906119388|5.3060528449921955 1.66
1 ✅ Good T cell 13.575245113590226 2.07
2 ⚠️ ? Monocyte|Macrophage 14.798208690107241|8.829828211698532 1.68
3 ✅ Good B cell 13.794561862211818 4.00
4 ⚠️ ? Natural killer cell|T cell 9.334956049479073|7.343244933648183 1.27
5 ⚠️ ? Monocyte|Macrophage 13.918759306986406|10.037946625130374 1.39
6 ✅ Good Natural killer cell 15.30826245310867 3.40
7 ✅ Good Monocyte 10.787406042786724 2.18
8 ⚠️ ? T cell|CD8+ T cell 5.431184801334877|4.133502801071792 1.31
================================================================================
我们可以只查看注释质量较好的结果。
scsa.cell_auto_anno(adata,key='scsa_celltype_cellmarker')
...cell type added to scsa_celltype_cellmarker on obs of anndata
我们也可以使用 panglaodb 作为 target 来进行细胞类型注释。
scsa=ov.single.pySCSA(
adata=adata,
foldchange=1.5,
pvalue=0.01,
celltype='normal',
target='panglaodb',
tissue='All',
model_path='temp/pySCSA_2024_v1_plus.db'
)
res=scsa.cell_anno(
clustertype='leiden',
cluster='all',
rank_rep=True
)
ranking genes
finished (0:00:00)
...Auto annotate cell
🔍 Version V2.2 [2024/12/18]
📊 DB load: GO_items:47347, Human_GO:3, Mouse_GO:3,
CellMarkers:82887, CancerSEA:1574, PanglaoDB:24223
Ensembl_HGNC:61541, Ensembl_Mouse:55414
<omicverse.single._SCSA.Annotator object at 0x14c448050>
🔍 Version V2.2 [2024/12/18]
📊 DB load: GO_items:47347, Human_GO:3, Mouse_GO:3,
CellMarkers:82887, CancerSEA:1574, PanglaoDB:24223
Ensembl_HGNC:61541, Ensembl_Mouse:55414
📦 Load markers: 70276
============================================================
🔬 Analyzing 9 clusters...
============================================================
[1/9] Cluster 0 │ 75 genes │ 632 other genes
[2/9] Cluster 1 │ 154 genes │ 602 other genes
[3/9] Cluster 2 │ 581 genes │ 572 other genes
[4/9] Cluster 3 │ 128 genes │ 592 other genes
[5/9] Cluster 4 │ 81 genes │ 635 other genes
[6/9] Cluster 5 │ 908 genes │ 538 other genes
[7/9] Cluster 6 │ 256 genes │ 586 other genes
[8/9] Cluster 7 │ 52 genes │ 645 other genes
[9/9] Cluster 8 │ 5 genes │ 645 other genes
============================================================
✅ Cluster analysis completed! (9/9 processed)
============================================================
================================================================================
📋 Cell Type Annotation Results
================================================================================
Cluster Type Cell Type Score Times
--------------------------------------------------------------------------------
0 ⚠️ ? T Cells|T Memory Cells 3.7202138087000143|3.3571403840625624 1.11
1 ⚠️ ? T Cells|T Memory Cells 3.5389401028805043|3.109624332162554 1.14
2 ⚠️ ? Monocytes|Alveolar Macrophages 3.6648210820925704|2.9377520436871687 1.25
3 ⚠️ ? B Cells Naive|B Cells Memory 4.335481613464625|3.9591672199193098 1.10
4 ⚠️ ? NK Cells|T Cells 2.9343417206491886|2.5083417903196352 1.17
5 ⚠️ ? Monocytes|Macrophages 3.762558876283017|2.8175042671102175 1.34
6 ⚠️ ? NK Cells|Gamma Delta T Cells 4.052418431477111|2.8660094064808934 1.41
7 ⚠️ ? Monocytes|Alveolar Macrophages 2.597715597444312|2.1244779821849584 1.22
8 ⚠️ ? Decidual Cells|NK Cells 1.629486719474794|1.629486719474794 1.00
================================================================================
我们同样可以只查看注释质量较好的结果。
scsa.cell_anno_print()
Cluster:0 Cell_type:T Cells|T Memory Cells Z-score:3.72|3.357
Cluster:1 Cell_type:T Cells|T Memory Cells Z-score:3.539|3.11
Cluster:2 Cell_type:Monocytes|Alveolar Macrophages Z-score:3.665|2.938
Cluster:3 Cell_type:B Cells Naive|B Cells Memory Z-score:4.335|3.959
Cluster:4 Cell_type:NK Cells|T Cells Z-score:2.934|2.508
Cluster:5 Cell_type:Monocytes|Macrophages Z-score:3.763|2.818
Cluster:6 Cell_type:NK Cells|Gamma Delta T Cells Z-score:4.052|2.866
Cluster:7 Cell_type:Monocytes|Alveolar Macrophages Z-score:2.598|2.124
Cluster:8 Cell_type:Decidual Cells|NK Cells Z-score:1.629|1.629
scsa.cell_auto_anno(adata,key='scsa_celltype_panglaodb')
...cell type added to scsa_celltype_panglaodb on obs of anndata
这里介绍降维可视化函数 ov.utils.embedding。它与 scanpy.pl.embedding 类似,不同之处在于当设置 frameon='small' 时,坐标轴会缩放到左下角,颜色条会缩放到右下角。
adata: anndata 对象
basis: 存储在
adata.obsm中、用于展示的嵌入坐标color: 需要可视化的 obs/var 信息
legend_loc: 图例位置;若设为
None,会显示在右侧frameon: 可以设置为
small、False或Nonelegend_fontoutline: 图例文字描边宽度
palette: 用于指定不同类别的颜色。除了 omicverse 的默认调色板外,也可以像下面这样直接传入颜色字典,以获得更稳定的显示效果。
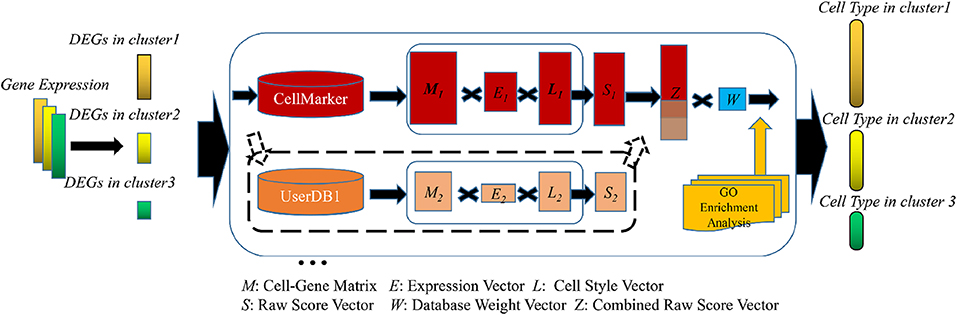
ov.utils.embedding(
adata,
basis='X_mde',
color=['leiden','scsa_celltype_cellmarker','scsa_celltype_panglaodb'],
legend_loc='on data',
frameon='small',
legend_fontoutline=2,
palette=ov.utils.palette()[9:],
)
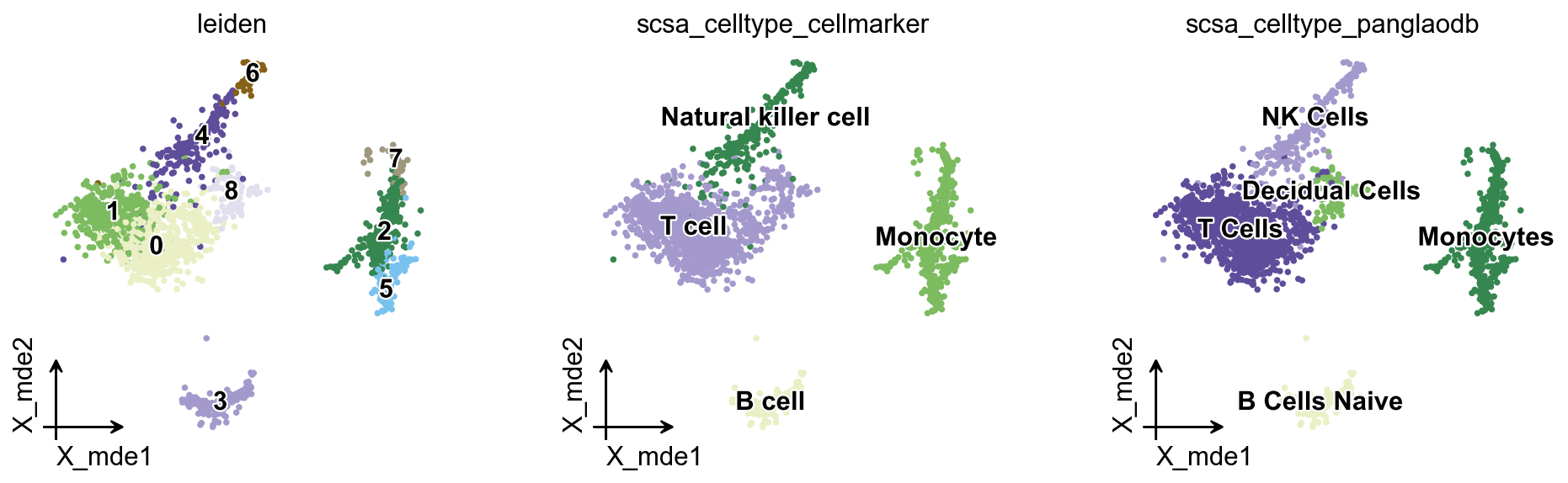
如果你想绘制细胞类型比例的堆叠柱状图,首先需要使用 ov.utils.embedding 对待比较的分组进行着色,然后再使用 ov.utils.plot_cellproportion 指定要展示的分组,即可查看不同组中的细胞比例。
# 随机将前 1000 个细胞标记为 B 组,其余细胞标记为 A 组
adata.obs['group']='A'
adata.obs.loc[adata.obs.index[:1000],'group']='B'
# 着色展示
ov.utils.embedding(
adata,
basis='X_mde',
color=['group'],
frameon='small',
legend_fontoutline=2,
palette={'A': '#F0C3C3', 'B': '#CB3E35'},
)
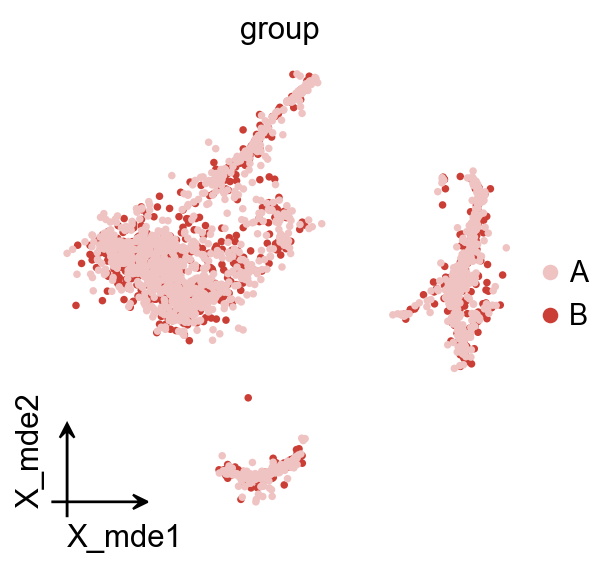
ov.utils.plot_cellproportion(
adata=adata,
celltype_clusters='scsa_celltype_cellmarker',
visual_clusters='group',
visual_name='group',
figsize=(2,4)
)
(<Figure size 160x320 with 1 Axes>,
<Axes: xlabel='group', ylabel='Cells per Stage'>)
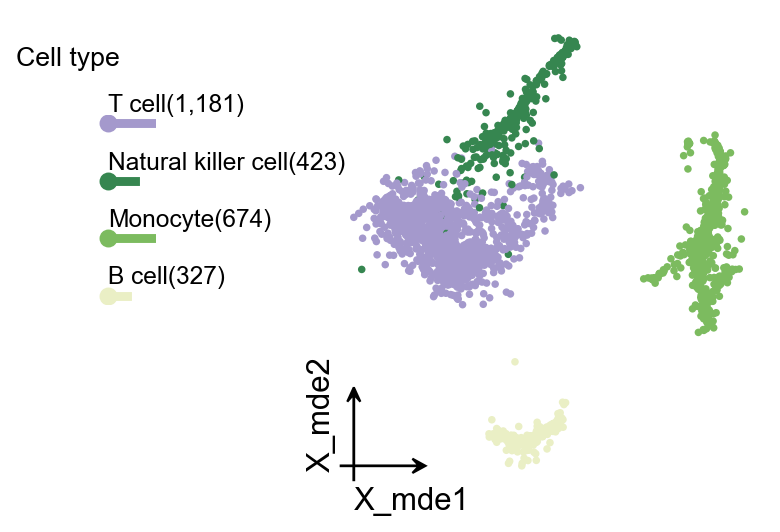
当然,我们还提供了另一种缩略式的嵌入图可视化函数 ov.utils.plot_embedding_celltype。
ov.utils.plot_embedding_celltype(
adata,
figsize=None,
basis='X_mde',
celltype_key='scsa_celltype_cellmarker',
title='Cell type',
celltype_range=(2,6),
embedding_range=(4,10)
)
(<Figure size 480x320 with 2 Axes>,
[<Axes: xlabel='X_mde1', ylabel='X_mde2'>, <Axes: >])
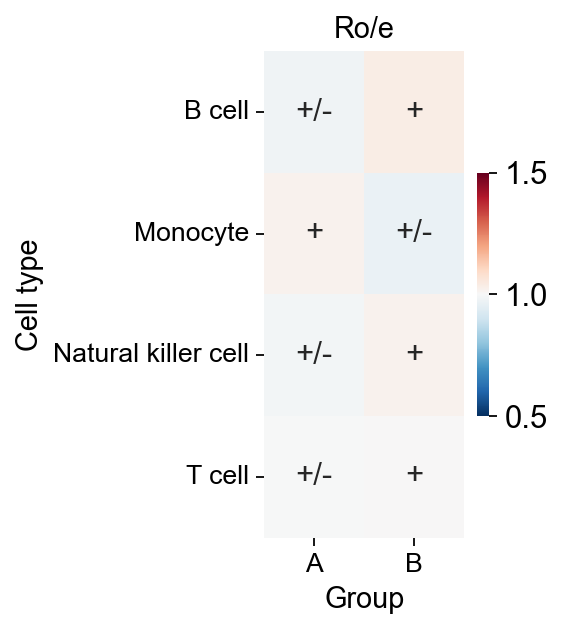
我们计算了不同组织中每个簇的观测细胞数与期望细胞数之比(Ro/e),以量化每个簇的组织偏好性(Guo et al., 2018; Zhang et al., 2018)。细胞簇与组织组合对应的期望细胞数由卡方检验得到。当某个簇在特定组织中的 Ro/e > 1 时,可以认为该簇在该组织中富集。
Ro/e 相关函数由 Haihao Zhang 编写。
roe=ov.utils.roe(
adata,
sample_key='group',
cell_type_key='scsa_celltype_cellmarker'
)
chi2: 0.8694929746430213, dof: 3, pvalue: 0.8327829060823263
P-value is greater than 0.05, there is no statistical significance
import seaborn as sns
import matplotlib.pyplot as plt
fig, ax = plt.subplots(figsize=(2,4))
transformed_roe = roe.copy()
transformed_roe = transformed_roe.applymap(
lambda x: '+++' if x >= 2 else ('++' if x >= 1.5 else ('+' if x >= 1 else '+/-')))
sns.heatmap(
roe,
annot=transformed_roe,
cmap='RdBu_r',
fmt='',
cbar=True,
ax=ax,
vmin=0.5,
vmax=1.5,
cbar_kws={'shrink':0.5}
)
plt.xticks(fontsize=12)
plt.yticks(fontsize=12)
plt.xlabel('Group',fontsize=13)
plt.ylabel('Cell type',fontsize=13)
plt.title('Ro/e',fontsize=13)
Text(0.5, 1.0, 'Ro/e')
手动细胞注释#
为了比较自动注释结果的准确性,这里我们将使用 marker genes 对各个簇进行手动注释,并将手动注释结果与 pySCSA 的结果进行对比。
首先需要准备一个 marker 基因字典。
res_marker_dict={
'Megakaryocyte':['ITGA2B','ITGB3'],
'Dendritic cell':['CLEC10A','IDO1'],
'Monocyte' :['S100A8','S100A9','LST1',],
'Macrophage':['CSF1R','CD68'],
'B cell':['MS4A1','CD79A','MZB1',],
'NK/NKT cell':['GNLY','KLRD1'],
'CD8+T cell':['CD8A','CD8B'],
'Treg':['CD4','CD40LG','IL7R','FOXP3','IL2RA'],
'CD4+T cell':['PTPRC','CD3D','CD3E'],
}
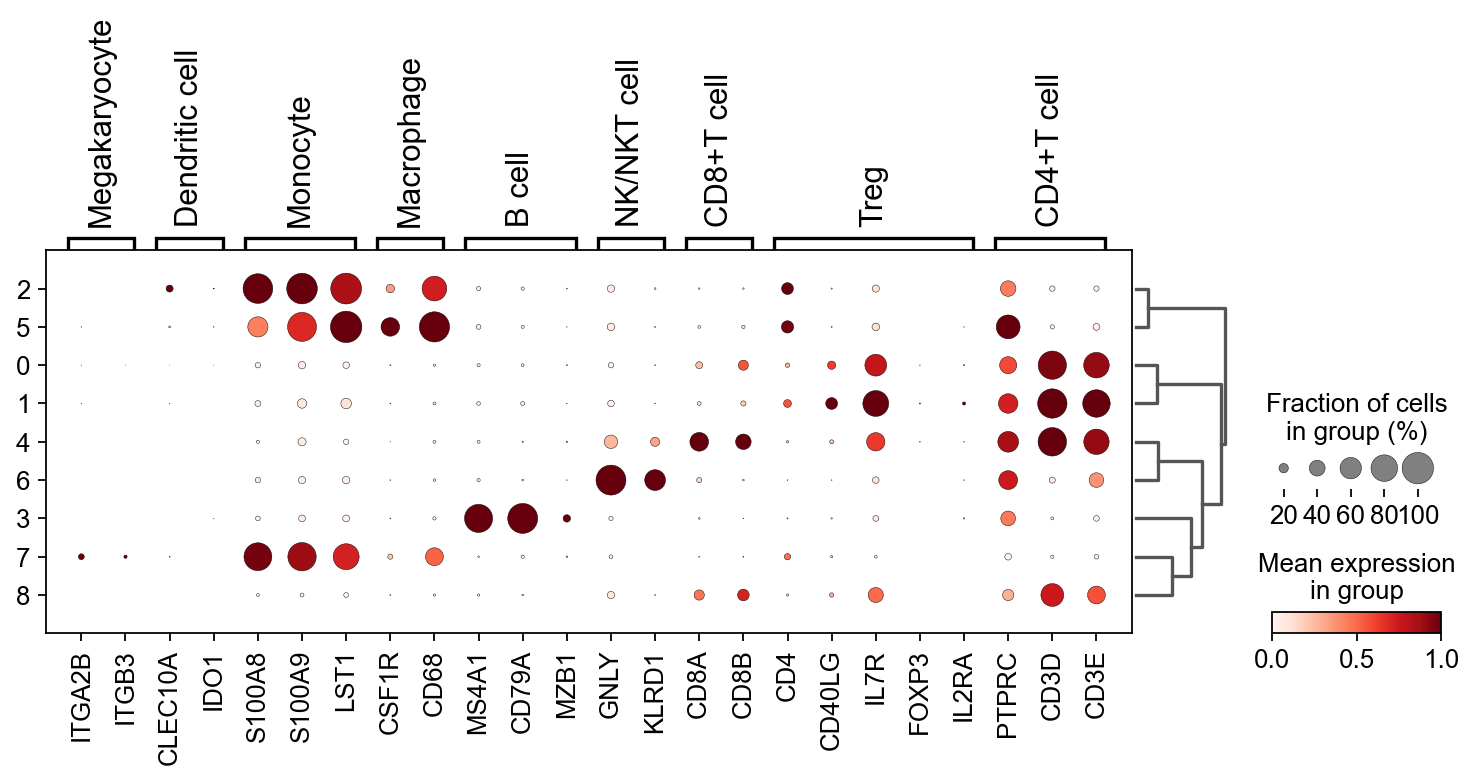
接着,我们计算每个簇中 marker genes 的表达水平及其表达比例。
sc.tl.dendrogram(adata,'leiden')
sc.pl.dotplot(
adata,
res_marker_dict,
'leiden',
dendrogram=True,
standard_scale='var'
)
using 'X_pca' with n_pcs = 50
Storing dendrogram info using `.uns['dendrogram_leiden']`
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different.
categories: 0, 1, 2, etc.
var_group_labels: Megakaryocyte, Dendritic cell, Monocyte, etc.
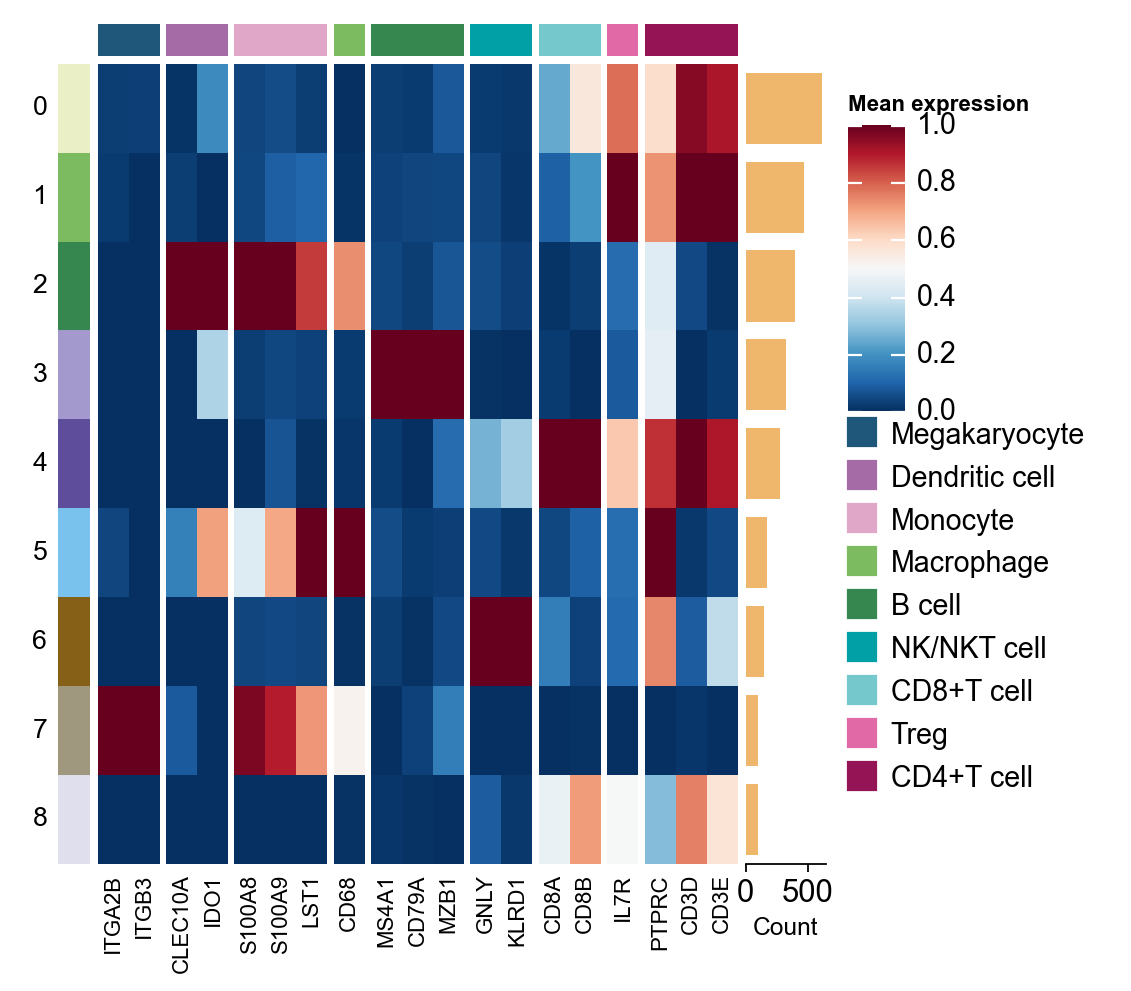
我们也可以使用基于 Marsilea 的新分组热图来展示同一批 marker genes。
marker_genes_heatmap = {k: v for k, v in res_marker_dict.items() if len(v) > 0}
h = ov.pl.group_heatmap(
adata,
var_names=marker_genes_heatmap,
groupby='leiden',
figsize=(4, 5),
standard_scale='var',
cmap='RdBu_r',
border=False,
show=False
)
基于 dotplot 的结果,我们使用 ov.single.scanpy_cellanno_from_dict 为每个簇指定名称。
# 创建簇到注释标签的映射字典
cluster2annotation = {
'0': 'T cell',
'1': 'T cell',
'2': 'Monocyte', # Germ-cell(Oid)
'3': 'B cell', # Germ-cell(Oid)
'4': 'T cell',
'5': 'Macrophage',
'6': 'NKT cells',
'7': 'Monocyte',
'8': 'T cell',
'9': 'Dendritic cell',
'10':'Megakaryocyte',
}
ov.single.scanpy_cellanno_from_dict(
adata,anno_dict=cluster2annotation,
clustertype='leiden'
)
...cell type added to major_celltype on obs of anndata
比较 pySCSA 与手动注释#
可以看到,自动注释结果与手动注释结果几乎一致,唯一的差异主要出现在 monocyte 与 macrophage 的区分上。不过在前面的自动注释结果中,pySCSA 也给出了 monocyte|macrophage 这样的候选结果,因此可以认为 pySCSA 在 pbmc3k 数据上的表现仍然较好。
ov.utils.embedding(
adata,
basis='X_mde',
color=['leiden','major_celltype','scsa_celltype_cellmarker'],
legend_loc='on data',
frameon='small',
legend_fontoutline=2,
palette=ov.utils.palette()[9:],
)
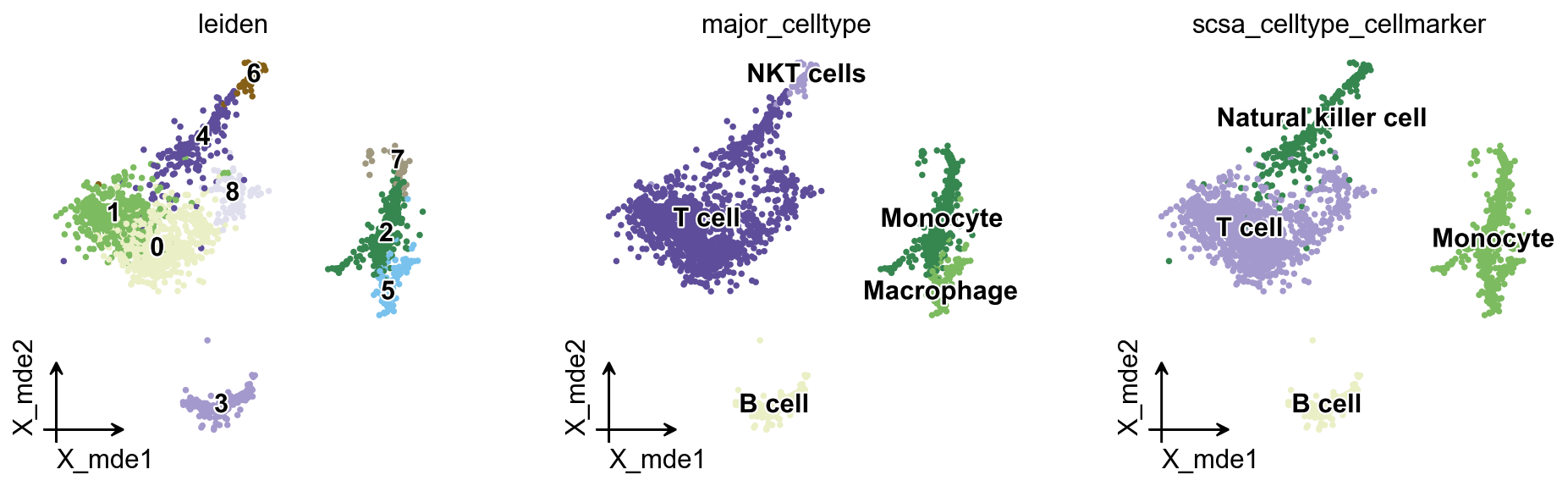
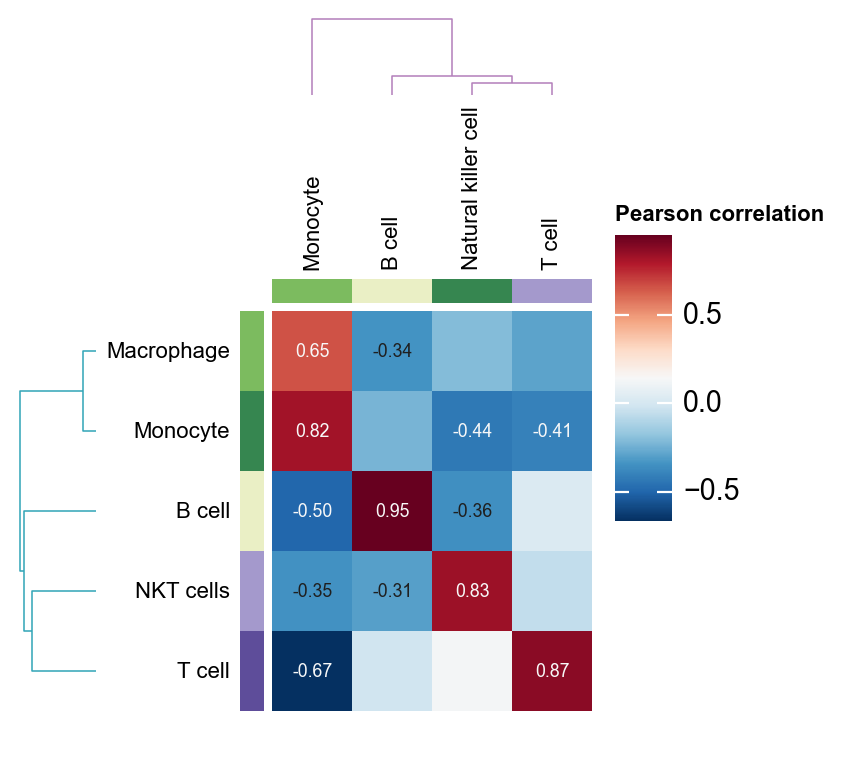
我们也可以使用 ov.pl.cell_cor_heatmap 来比较手动注释结果与 pySCSA 自动注释结果之间的表达相似性。
cell_cor_h = ov.pl.cell_cor_heatmap(
adata,
group_by='major_celltype',
ref_adata=adata,
ref_group_by='scsa_celltype_cellmarker',
method='pearson',
standard_scale='var',
cmap='RdBu_r',
figsize=(2, 2.5),
row_cluster=True,
col_cluster=True,
show_values=True,
value_cutoff=0.3,
border=False,
show=False,
)
我们可以使用 get_celltype_marker 获取每种细胞类型对应的 marker genes。
marker_dict=ov.single.get_celltype_marker(adata,clustertype='scsa_celltype_cellmarker')
marker_dict.keys()
...get cell type marker
ranking genes
finished (0:00:00)
dict_keys(['B cell', 'Monocyte', 'Natural killer cell', 'T cell'])
marker_dict['B cell']
['CD37',
'HLA-DPB1',
'HLA-DQA1',
'CD74',
'MS4A1',
'HLA-DRB1',
'HLA-DRA',
'CD79B',
'HLA-DQB1',
'CD79A']
数据库中的组织名称#
如果需要对特定组织中的细胞类型进行注释,可以使用 get_model_tissue 查询数据库中可用的组织名称。
scsa.get_model_tissue()
🔍 Version V2.2 [2024/12/18]
📊 DB load: GO_items:47347, Human_GO:3, Mouse_GO:3,
CellMarkers:82887, CancerSEA:1574, PanglaoDB:24223
Ensembl_HGNC:61541, Ensembl_Mouse:55414
########################################################################################################################
------------------------------------------------------------------------------------------------------------------------
Species:Human Num:298
------------------------------------------------------------------------------------------------------------------------
1: Abdomen 2: Abdominal adipose tissue 3: Abdominal fat pad
4: Acinus 5: Adipose tissue 6: Adrenal gland
7: Adventitia 8: Airway 9: Airway epithelium
10: Allocortex 11: Alveolus 12: Amniotic fluid
13: Amniotic membrane 14: Ampullary 15: Anogenital tract
16: Antecubital vein 17: Anterior cruciate ligament 18: Anterior presomitic mesoderm
19: Aorta 20: Aortic valve 21: Artery
22: Arthrosis 23: Articular Cartilage 24: Ascites
25: Ascitic fluid 26: Atrium 27: Basal airway
28: Basilar membrane 29: Beige Fat 30: Bile duct
31: Biliary tract 32: Bladder 33: Blood
34: Blood vessel 35: Bone 36: Bone marrow
37: Brain 38: Breast 39: Bronchial vessel
40: Bronchiole 41: Bronchoalveolar lavage 42: Bronchoalveolar system
43: Bronchus 44: Brown adipose tissue 45: Calvaria
46: Capillary 47: Cardiac atrium 48: Cardiovascular system
49: Carotid artery 50: Carotid plaque 51: Cartilage
52: Caudal cortex 53: Caudal forebrain 54: Caudal ganglionic eminence
55: Cavernosum 56: Central amygdala 57: Central nervous system
58: Cerebellum 59: Cerebral organoid 60: Cerebrospinal fluid
61: Cervix 62: Choriocapillaris 63: Chorionic villi
64: Chorionic villus 65: Choroid 66: Choroid plexus
67: Colon 68: Colon epithelium 69: Colorectum
70: Cornea 71: Corneal endothelium 72: Corneal epithelium
73: Coronary artery 74: Corpus callosum 75: Corpus luteum
76: Cortex 77: Cortical layer 78: Cortical thymus
79: Decidua 80: Deciduous tooth 81: Dental pulp
82: Dermis 83: Diencephalon 84: Distal airway
85: Dorsal forebrain 86: Dorsal root ganglion 87: Dorsolateral prefrontal cortex
88: Ductal tissue 89: Duodenum 90: Ectocervix
91: Ectoderm 92: Embryo 93: Embryoid body
94: Embryonic Kidney 95: Embryonic brain 96: Embryonic heart
97: Embryonic prefrontal cortex 98: Embryonic stem cell 99: Endocardium
100: Endocrine 101: Endoderm 102: Endometrium
103: Endometrium stroma 104: Entorhinal cortex 105: Epidermis
106: Epithelium 107: Esophageal 108: Esophagus
109: Eye 110: Fat pad 111: Fetal brain
112: Fetal gonad 113: Fetal heart 114: Fetal ileums
115: Fetal kidney 116: Fetal liver 117: Fetal lung
118: Fetal thymus 119: Fetal umbilical cord 120: Fetus
121: Foreskin 122: Frontal cortex 123: Fundic gland
124: Gall bladder 125: Gastric corpus 126: Gastric epithelium
127: Gastric gland 128: Gastrointestinal tract 129: Germ
130: Germinal center 131: Gingiva 132: Gonad
133: Gut 134: Hair follicle 135: Head
136: Head and neck 137: Heart 138: Heart muscle
139: Hippocampus 140: Ileum 141: Iliac crest
142: Inferior colliculus 143: Intervertebral disc 144: Intestinal crypt
145: Intestine 146: Intrahepatic cholangio 147: Jejunum
148: Kidney 149: Lacrimal gland 150: Large Intestine
151: Large intestine 152: Larynx 153: Lateral ganglionic eminence
154: Left lobe 155: Ligament 156: Limb bud
157: Limbal epithelium 158: Liver 159: Lumbar vertebra
160: Lung 161: Lymph 162: Lymph node
163: Lymphatic vessel 164: Lymphoid tissue 165: Malignant pleural effusion
166: Mammary epithelium 167: Mammary gland 168: Medial ganglionic eminence
169: Medullary thymus 170: Meniscus 171: Mesenchyme
172: Mesoblast 173: Mesoderm 174: Microvascular endothelium
175: Microvessel 176: Midbrain 177: Middle temporal gyrus
178: Milk 179: Molar 180: Muscle
181: Myenteric plexus 182: Myocardium 183: Myometrium
184: Nasal concha 185: Nasal epithelium 186: Nasal mucosa
187: Nasal polyp 188: Nasopharyngeal mucosa 189: Nasopharynx
190: Neck 191: Neocortex 192: Nerve
193: Nose 194: Nucleus pulposus 195: Olfactory neuroepithelium
196: Omentum 197: Optic nerve 198: Oral cavity
199: Oral mucosa 200: Osteoarthritic cartilage 201: Ovarian cortex
202: Ovarian follicle 203: Ovary 204: Oviduct
205: Palatine tonsil 206: Pancreas 207: Pancreatic acinar tissue
208: Pancreatic duct 209: Pancreatic islet 210: Periodontal ligament
211: Periodontium 212: Periosteum 213: Peripheral blood
214: Peritoneal fluid 215: Peritoneum 216: Pituitary
217: Pituitary gland 218: Placenta 219: Plasma
220: Pleura 221: Pluripotent stem cell 222: Polyp
223: Posterior fossa 224: Posterior presomitic mesoderm 225: Prefrontal cortex
226: Premolar 227: Presomitic mesoderm 228: Primitive streak
229: Prostate 230: Pulmonary arteriy 231: Pyloric gland
232: Rectum 233: Renal glomerulus 234: Respiratory tract
235: Retina 236: Retinal organoid 237: Retinal pigment epithelium
238: Right ventricle 239: Saliva 240: Salivary gland
241: Scalp 242: Sclerocorneal tissue 243: Seminal plasma
244: Septum transversum 245: Serum 246: Sinonasal mucosa
247: Sinus tissue 248: Skeletal muscle 249: Skin
250: Small intestine 251: Soft tissue 252: Sperm
253: Spinal cord 254: Spleen 255: Sputum
256: Stomach 257: Subcutaneous adipose tissue 258: Submandibular gland
259: Subpallium 260: Subplate 261: Subventricular zone
262: Superior frontal gyrus 263: Sympathetic ganglion 264: Synovial fluid
265: Synovium 266: Taste bud 267: Tendon
268: Testis 269: Thalamus 270: Thymus
271: Thyroid 272: Tongue 273: Tonsil
274: Tooth 275: Trachea 276: Transformed artery
277: Trophoblast 278: Umbilical cord 279: Umbilical cord blood
280: Umbilical vein 281: Undefined 282: Urine
283: Urothelium 284: Uterine cervix 285: Uterus
286: Vagina 287: Vein 288: Venous blood
289: Ventral thalamus 290: Ventricular and atrial 291: Ventricular zone
292: Vessel 293: Visceral adipose tissue 294: Vocal cord
295: Vocal fold 296: White adipose tissue 297: White matter
########################################################################################################################