使用 scTour 进行轨迹推断#
这里以胰腺内分泌发育数据为例,使用原始 UMI counts 演示 scTour 的 latent-time 学习流程,以及基于 neural ordinary differential equation 的轨迹建模方式。
方法背景#
参考 scTour 官方文档 和原始 Genome Biology 论文,scTour 是一个深度学习框架,可以直接从 abundance matrix 中联合学习 latent representation、developmental pseudotime 和 vector field。
它的核心思路可以概括为:
先把细胞编码到能够表征发育结构的 latent space 中
在无需 spliced/unspliced counts 的前提下学习 pseudotime
用 neural ODE 风格的动力学模型描述连续的状态转变
利用学到的 latent dynamics 同时支持轨迹推断和细胞进展预测
因此,当我们希望使用神经网络式的 latent-time 模型,或者当前数据没有 RNA velocity 前处理输入时,scTour 会是一个很有吸引力的选择。
为什么这里使用胰腺数据?#
胰腺内分泌发育具有紧凑的连续进展过程和清晰的中间 endocrine 状态,因此很适合观察 scTour 是否能够恢复合理的 latent-time 排序与发育方向。
数据预处理#
这里我们以胰腺发育数据为例演示轨迹推断。
import scanpy as sc
import matplotlib.pyplot as plt
import warnings
warnings.filterwarnings("ignore", category=FutureWarning)
import omicverse as ov
ov.plot_set(font_path='Arial')
%load_ext autoreload
%autoreload 2
🔬 Starting plot initialization...
Using already downloaded Arial font from: /var/folders/rv/3jnfbs0d6r7d0c5bfj7ft5k00000gn/T/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.1.3rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
adata = ov.datasets.pancreatic_endocrinogenesis()
⚠️ File ./data/endocrinogenesis_day15.h5ad already exists
Loading data from ./data/endocrinogenesis_day15.h5ad
✅ Successfully loaded: 3696 cells × 27998 genes
adata = ov.pp.preprocess(adata, mode='shiftlog|pearson', n_HVGs=3000)
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features]
ov.pp.scale(adata)
ov.pp.pca(adata, layer='scaled', n_pcs=50)
🔍 [2026-05-12 15:49:23] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 17750/27998 genes are kept.
Among 17750 genes, 16426 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 1 highly-expressed genes from normalization computation
Excluded genes: ['Ghrl']
✅ Count Normalization Completed Successfully!
✓ Processed: 3,696 cells × 16,426 genes
✓ Runtime: 0.07s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 3,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 3,000 highly variable genes out of 16,426 total (18.3%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 0.46 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 0.5675s │
│ Shape: 3,696 x 27,998 -> 3,696 x 16,426 │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ REFERENCE_MANU │
│ │ ✚ _ov_provenance │
│ │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │ ✚ status │
│ │ ✚ status_args │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 3696x16426) │
│ │
╰────────────────────────────────────────────────────────────────────╯
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.2826s │
│ Shape: 3,696 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (array, 3696x3000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=50
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: ArrayView, shape: (3696, 3000), dtype: float64
🔧 PCA solver used: covariance_eigh
finished✅ (8.68s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 8.6833s │
│ Shape: 3,696 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ scaled|original|X_pca (array, 3696x50) │
│ │
╰────────────────────────────────────────────────────────────────────╯
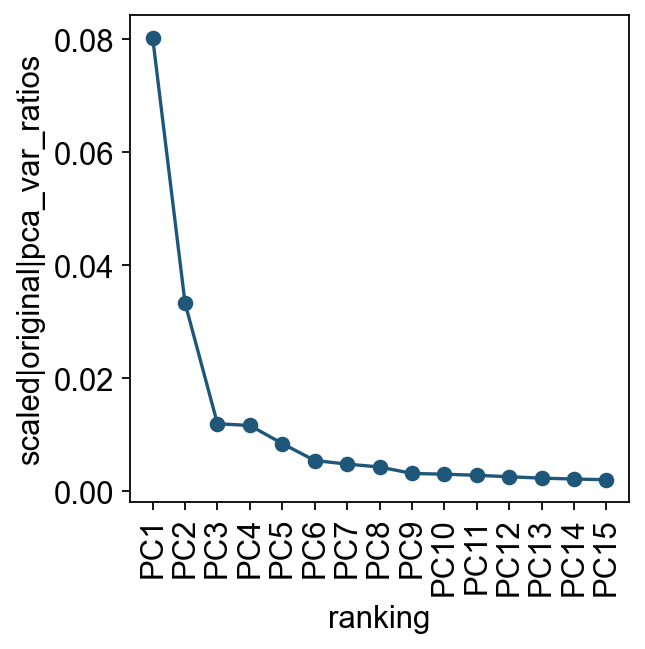
我们先查看各个主成分对总方差的贡献。这一步可以帮助判断后续构建细胞邻接关系时需要使用多少个 PC。实际分析中,通常只需要一个大致合理的 PC 数即可。
ov.utils.plot_pca_variance_ratio(adata, n_pcs=15)
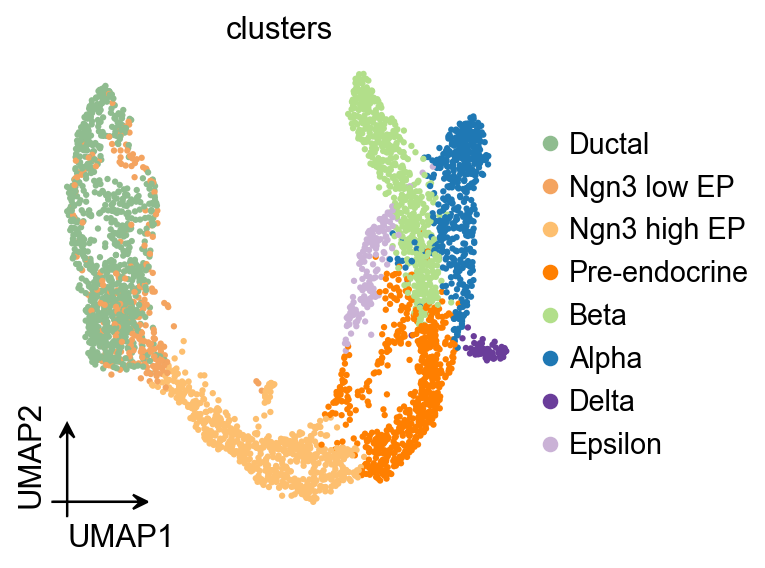
ov.pl.umap(
adata,
color='clusters'
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
scTour#
scTour 是一种用于解析单细胞组学数据中细胞动态过程的方法。
它把 developmental pseudotime、vector field 和 latent space 放在同一个框架中建模,从多个角度刻画发育过程。
接下来训练 scTour 模型。默认的 loss_mode 是 negative binomial conditioned likelihood (nb),需要把原始 UMI counts 放在 AnnData 的 .X 中。默认情况下,当细胞数少于 10,000 时使用 90% 的细胞训练;当细胞数超过 10,000 时使用 20% 的细胞训练。用户也可以通过 percent 参数调整训练细胞比例,例如 percent=0.6。
adata.X=adata.layers['counts'].copy()
sc.pp.calculate_qc_metrics(adata, percent_top=None, log1p=False, inplace=True)
Traj=ov.single.TrajInfer(
adata,basis='X_umap',
groupby='clusters',
use_rep='scaled|original|X_pca',
n_comps=50
)
Traj.inference(
method='sctour',
alpha_recon_lec=0.5,
alpha_recon_lode=0.5
)
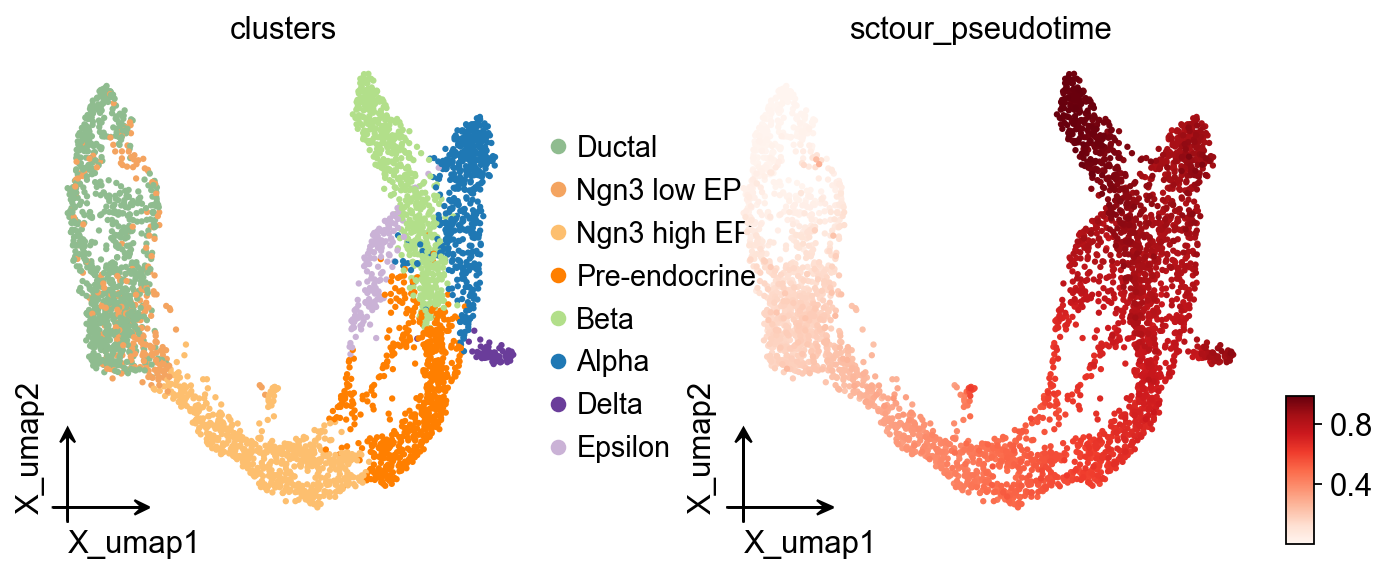
ov.pl.embedding(
adata,
basis='X_umap',
color=['clusters','sctour_pseudotime'],
frameon='small',
cmap='Reds'
)
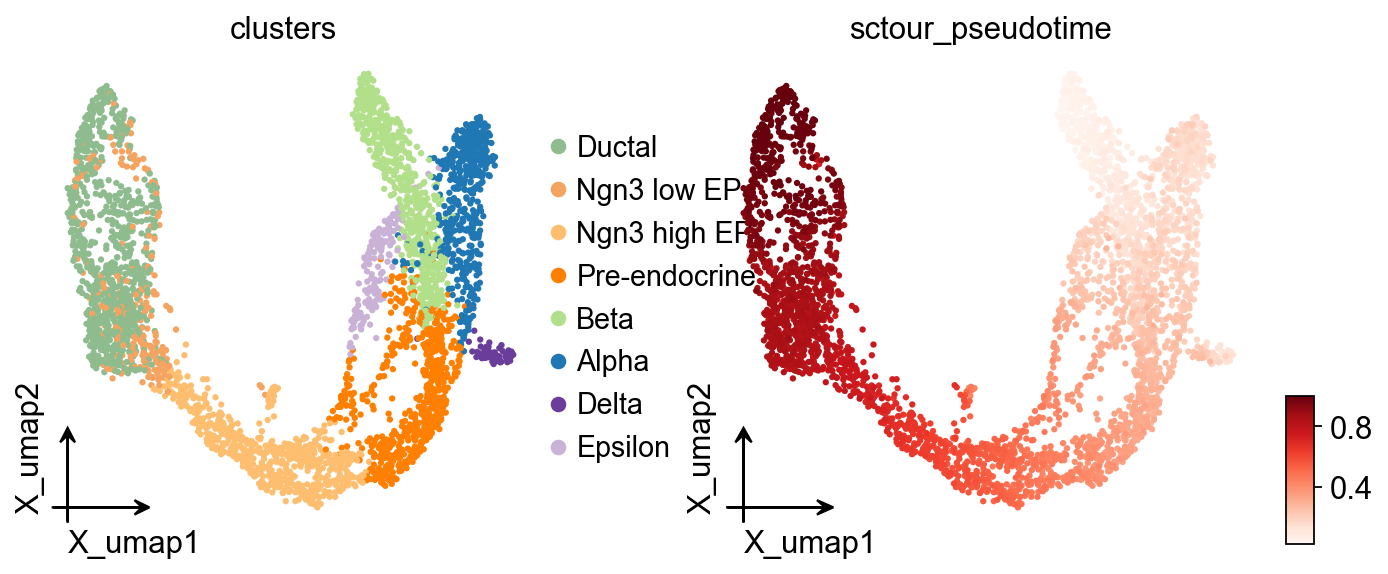
adata.obs['sctour_pseudotime']=1-adata.obs['sctour_pseudotime']
ov.pl.embedding(
adata,basis='X_umap',
color=['clusters','sctour_pseudotime'],
frameon='small',
cmap='Reds'
)
import os
os.makedirs('data', exist_ok=True)
adata.write('data/traj_tutorial.h5ad')
adata = ov.read('data/traj_tutorial.h5ad')
adata
AnnData object with n_obs × n_vars = 3696 × 3000
obs: 'clusters_coarse', 'clusters', 'S_score', 'G2M_score', 'n_genes_by_counts', 'total_counts', 'sctour_pseudotime'
var: 'highly_variable_genes', 'n_cells', 'percent_cells', 'robust', 'highly_variable_features', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts'
uns: 'REFERENCE_MANU', '_ov_provenance', 'clusters_coarse_colors', 'clusters_colors', 'clusters_sizes', 'day_colors', 'history_log', 'hvg', 'log1p', 'neighbors', 'paga', 'paga_graph', 'pca', 'scaled|original|cum_sum_eigenvalues', 'scaled|original|pca_var_ratios', 'status', 'status_args'
obsm: 'UMAP', 'X_TNODE', 'X_VF', 'X_pca', 'X_umap', 'scaled|original|X_pca'
varm: 'PCs', 'scaled|original|pca_loadings'
layers: 'counts', 'scaled', 'spliced', 'unspliced'
obsp: 'connectivities', 'distances'
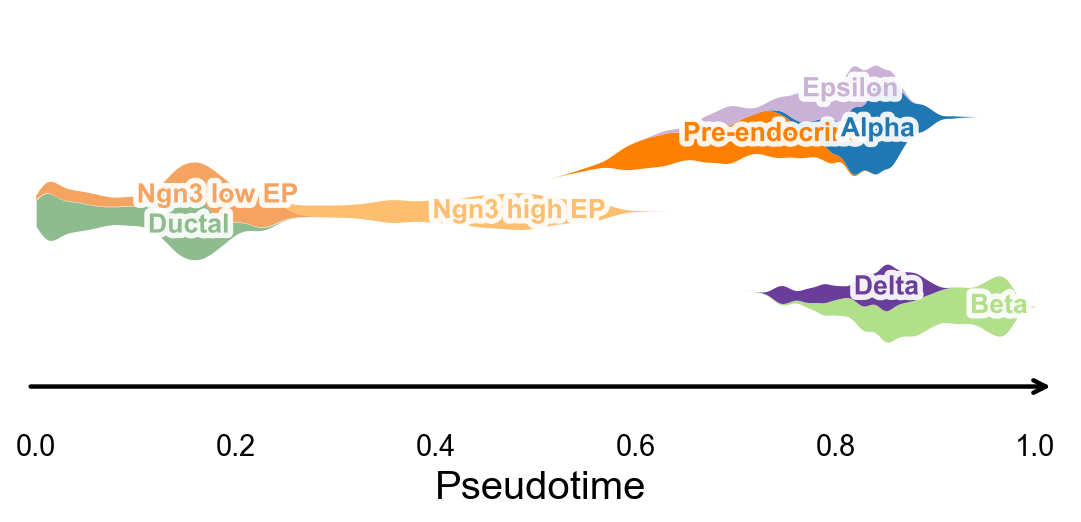
分支感知的伪时间流图#
ov.pl.branch_streamplot 只需要伪时间和细胞状态标签,因此也可以用于这个轨迹推断方法。图中 ribbon 的宽度表示某类细胞在伪时间上的富集位置,分叉的中心线则帮助观察不同 endocrine fate 在何处展开。
fig, ax = ov.pl.branch_streamplot(
adata,
group_key='clusters',
pseudotime_key='sctour_pseudotime',
show=False,
)
plt.show()
使用 dynamic_features / dynamic_trends 分析 scTour 伪时间#
同样的 GAM 趋势层也可以接到 sctour_pseudotime 上。这里先展示一张全局 marker 趋势图,并用 cluster 给原始散点着色;然后再比较晚期 Alpha / Beta 分支,展示分支相关的表达变化。
import numpy as np
import matplotlib.pyplot as plt
from IPython.display import display
sctour_genes = ['Sox9', 'Neurog3', 'Fev', 'Gcg', 'Arx', 'Pax4', 'Ins2', 'Pdx1', 'Sst', 'Hhex']
sctour_dyn = ov.single.dynamic_features(
adata,
genes=sctour_genes,
pseudotime='sctour_pseudotime',
use_raw=adata.raw is not None,
distribution='normal',
link='identity',
n_splines=8,
store_raw=True,
raw_obs_keys=['clusters'],
)
🔍 Dynamic feature analysis:
Views: 1 | Features: 10
Pseudotime: sctour_pseudotime
Stored raw obs keys: ['clusters']
Expression source: adata.raw
GAM: normal-identity | splines=8
✅ Dynamic feature analysis completed!
✓ Successful fits: 10/10
✓ Fitted rows: 2000
✓ Raw observations stored: 36960
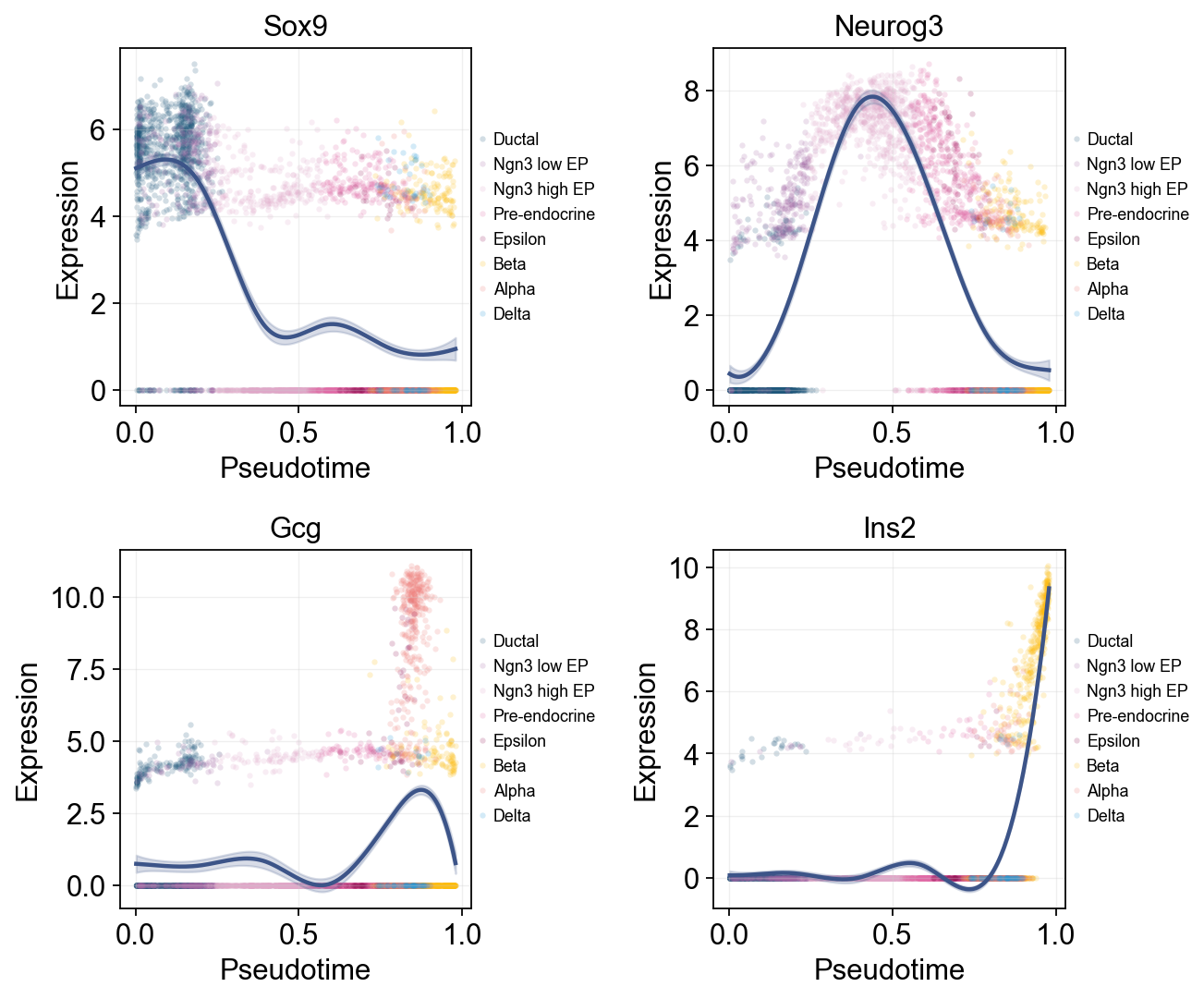
Single-line global trends#
这张图为每个基因只拟合一条全局趋势线,同时用细胞注释给原始散点着色。它适合区分“整体伪时间表达趋势”和“哪些细胞状态贡献了这些散点”。
fig, _ = ov.pl.dynamic_trends(
sctour_dyn,
genes=['Sox9', 'Neurog3', 'Gcg', 'Ins2'],
add_point=True,
point_color_by='clusters',
figsize=(5, 3.5),
legend_loc='right margin',
legend_fontsize=8,
ncols=2,
return_fig=True,
)
display(fig)
plt.close(fig)
🔍 Dynamic trend plotting:
Features: 4 | Groups: 1
compare_features=False | compare_groups=False
✅ Dynamic trend plotting completed!
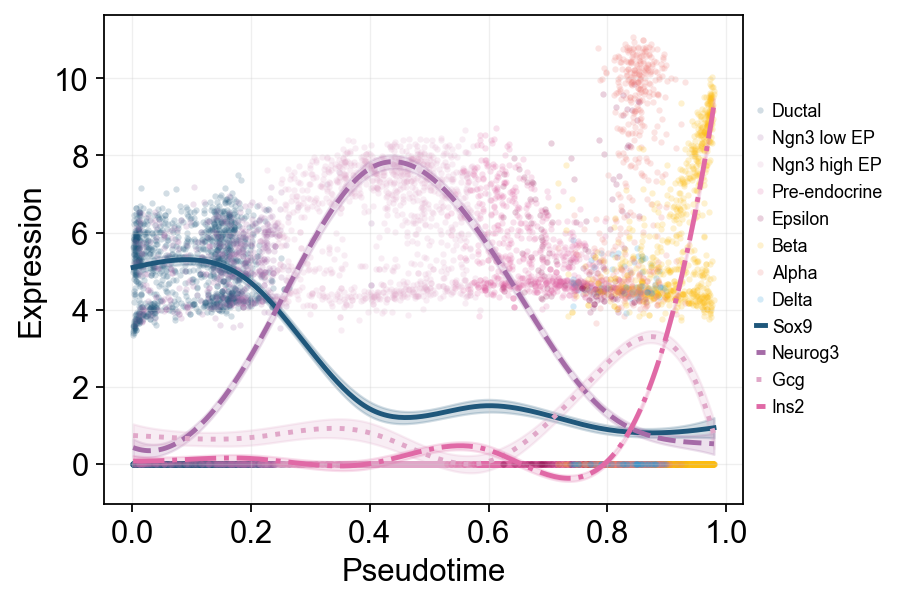
多标记基因趋势比较#
这里把多个 marker 的拟合曲线叠加在同一伪时间坐标中,方便直接比较不同程序的启动和衰减顺序。
fig, _ = ov.pl.dynamic_trends(
sctour_dyn,
genes=['Sox9', 'Neurog3', 'Gcg', 'Ins2'],
compare_features=True,
add_point=True,
point_color_by='clusters',
line_style_by='features',
figsize=(7, 4),
linewidth=2.2,
legend_loc='right margin',
legend_fontsize=8,
return_fig=True,
)
display(fig)
plt.close(fig)
🔍 Dynamic trend plotting:
Features: 4 | Groups: 1
compare_features=True | compare_groups=False
✅ Dynamic trend plotting completed!
branch_clusters = ['Alpha', 'Beta']
split_mask = adata.obs['clusters'].astype(str).isin(['Ngn3 high EP', 'Pre-endocrine'])
sctour_branch_dyn = ov.single.dynamic_features(
adata,
genes=['Gcg', 'Ins2', 'Pax4', 'Sox9'],
pseudotime='sctour_pseudotime',
groupby='clusters',
groups=branch_clusters,
use_raw=adata.raw is not None,
distribution='normal',
link='identity',
n_splines=8,
store_raw=True,
)
split_time = float(np.nanmedian(adata.obs.loc[split_mask, 'sctour_pseudotime'])) if split_mask.any() else float(np.nanmedian(adata.obs['sctour_pseudotime']))
🔍 Dynamic feature analysis:
Views: 2 | Features: 4
Pseudotime: sctour_pseudotime
Grouping: clusters
Expression source: adata.raw
GAM: normal-identity | splines=8
✅ Dynamic feature analysis completed!
✓ Successful fits: 8/8
✓ Fitted rows: 1600
✓ Raw observations stored: 4288
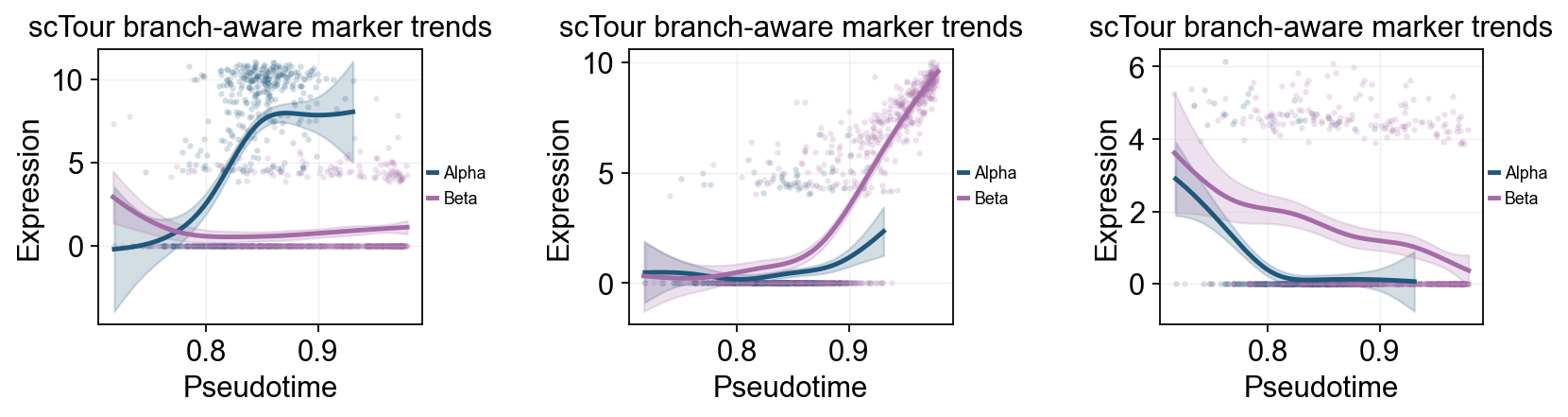
fig, _ = ov.pl.dynamic_trends(
sctour_branch_dyn,
genes=['Gcg', 'Ins2', 'Pax4'],
compare_groups=True,
split_time=split_time,
shared_trunk=True,
add_point=True,
point_color_by='group',
figsize=(4.2, 3),
ncols=3,
linewidth=2.2,
legend_loc='right margin',
legend_fontsize=8,
title='scTour branch-aware marker trends',
return_fig=True,
)
display(fig)
plt.close(fig)
🔍 Dynamic trend plotting:
Features: 3 | Groups: 2
compare_features=False | compare_groups=True
✅ Dynamic trend plotting completed!
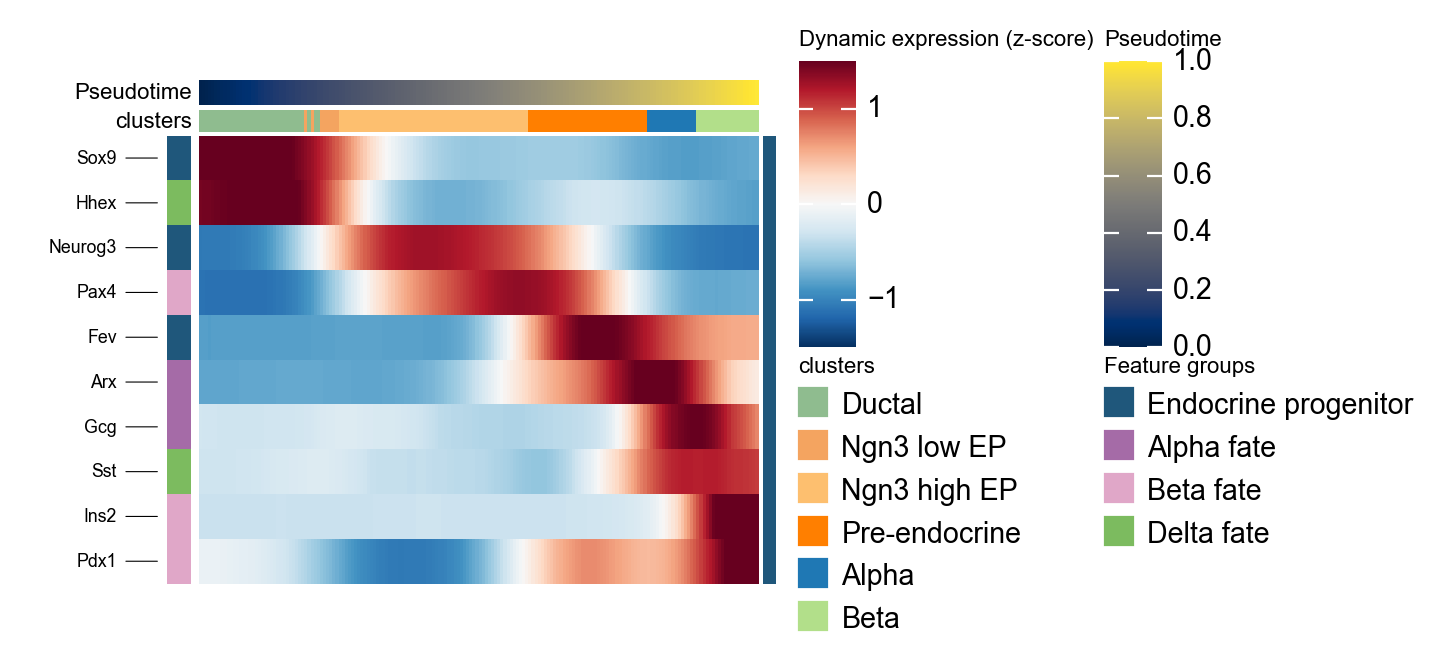
使用 dynamic_heatmap 概括 scTour 标记基因程序#
ov.pl.dynamic_heatmap 可以把多组 marker program 压缩成一张按伪时间排序的热图,用来检查 progenitor、Alpha、Beta 和 Delta 程序是否沿 scTour 伪时间按预期顺序启动。
sctour_marker = {
'Endocrine progenitor': ['Sox9', 'Neurog3', 'Fev'],
'Alpha fate': ['Gcg', 'Arx'],
'Beta fate': ['Pax4', 'Ins2', 'Pdx1'],
'Delta fate': ['Sst', 'Hhex'],
}
g = ov.pl.dynamic_heatmap(
adata,
var_names=sctour_marker,
pseudotime='sctour_pseudotime',
use_raw=adata.raw is not None,
use_cell_columns=False,
cell_annotation='clusters',
cell_bins=180,
smooth_window=17,
fitted_window=31,
figsize=(5, 4),
standard_scale='var',
cmap='RdBu_r',
use_fitted=True,
border=False,
show=False,
)
🔍 Dynamic heatmap:
Candidate features: 10
Pseudotime: sctour_pseudotime
Cell annotation: clusters
use_fitted=True | cell_bins=180 | cmap=RdBu_r
✅ Dynamic heatmap completed!
✓ Matrix shape: 10 features × 180 columns