使用 Slingshot 进行轨迹推断#
这里以胰腺内分泌发育数据为例,演示 Slingshot 的 lineage 拟合、伪时间可视化,以及带 lineage 信息的动态趋势总结。
方法背景#
参考 Bioconductor 的 Slingshot vignette 和原始 [BMC Genomics 论文](https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-018-4772-0
),Slingshot 将 cluster 拓扑和光滑曲线拟合结合起来进行轨迹推断。
它的核心思路可以概括为:
先在低维空间中对细胞进行聚类
用 minimum spanning tree 连接 cluster,恢复全局 lineage 结构
在这些 cluster 上拟合 simultaneous principal curves
为每条 lineage 计算 lineage-specific pseudotime,同时允许多个 lineage 共享早期主干
这种设计很适合“在 cluster 层面更容易解释拓扑结构”的发育体系。
为什么这里使用胰腺数据?#
胰腺数据具有紧凑而清晰的分支结构,既有明显的 endocrine 主干,也有多个终末细胞状态,因此很适合展示 Slingshot 的 lineage 拟合和后续的 lineage-aware 动态趋势总结。
数据预处理#
这里我们以胰腺发育数据为例演示轨迹推断。
import scanpy as sc
import matplotlib.pyplot as plt
import warnings
warnings.filterwarnings("ignore", category=FutureWarning)
import omicverse as ov
ov.plot_set(font_path='Arial')
%load_ext autoreload
%autoreload 2
🔬 Starting plot initialization...
Using already downloaded Arial font from: /var/folders/rv/3jnfbs0d6r7d0c5bfj7ft5k00000gn/T/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
adata = ov.datasets.pancreatic_endocrinogenesis()
⚠️ File ./data/endocrinogenesis_day15.h5ad already exists
Loading data from ./data/endocrinogenesis_day15.h5ad
✅ Successfully loaded: 3696 cells × 27998 genes
adata = ov.pp.preprocess(adata, mode='shiftlog|pearson', n_HVGs=3000)
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features]
ov.pp.scale(adata)
ov.pp.pca(adata, layer='scaled', n_pcs=50)
🔍 [2026-05-22 18:42:49] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 17750/27998 genes are kept.
Among 17750 genes, 16426 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 1 highly-expressed genes from normalization computation
Excluded genes: ['Ghrl']
✅ Count Normalization Completed Successfully!
✓ Processed: 3,696 cells × 16,426 genes
✓ Runtime: 0.11s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 3,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 3,000 highly variable genes out of 16,426 total (18.3%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 0.94 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 1.0725s │
│ Shape: 3,696 x 27,998 -> 3,696 x 16,426 │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ REFERENCE_MANU │
│ │ ✚ _ov_provenance │
│ │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │ ✚ status │
│ │ ✚ status_args │
│ │
│ ● LAYERS │ ✚ counts (sparse matrix, 3696x16426) │
│ │
╰────────────────────────────────────────────────────────────────────╯
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.41s │
│ Shape: 3,696 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (array, 3696x3000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=50
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: ArrayView, shape: (3696, 3000), dtype: float64
🔧 PCA solver used: covariance_eigh
finished✅ (9.92s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 9.9253s │
│ Shape: 3,696 x 3,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ scaled|original|X_pca (array, 3696x50) │
│ │
╰────────────────────────────────────────────────────────────────────╯
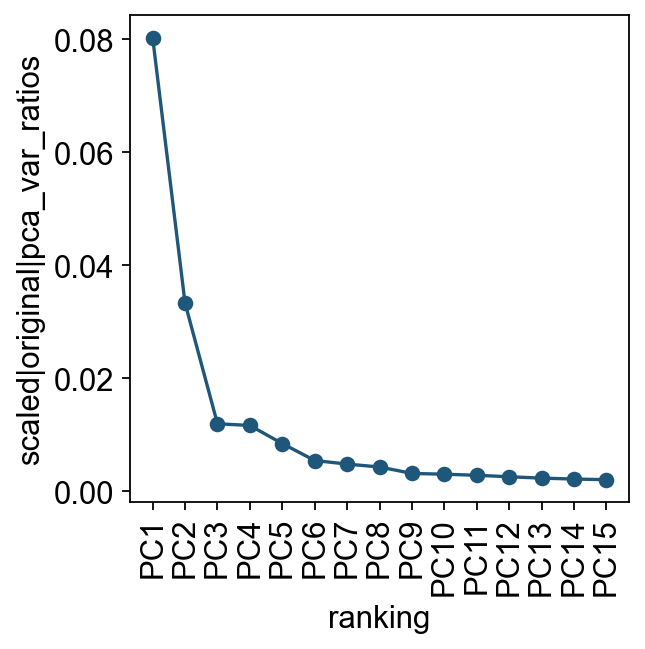
我们先查看各个主成分对总方差的贡献。这一步可以帮助判断后续构建细胞邻接关系时需要使用多少个 PC。实际分析中,通常只需要一个大致合理的 PC 数即可。
ov.utils.plot_pca_variance_ratio(adata, n_pcs=15)
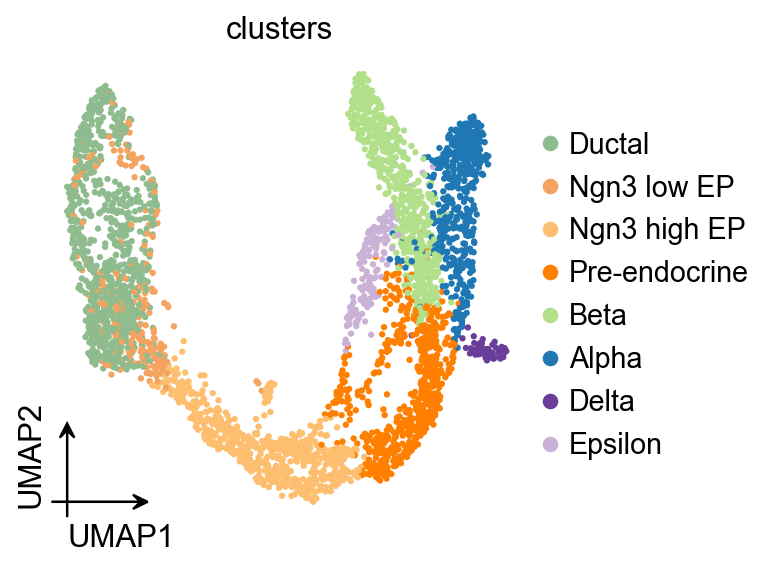
ov.pl.umap(
adata,
color='clusters'
)
X_umap converted to UMAP to visualize and saved to adata.obsm['UMAP']
if you want to use X_umap, please set convert=False
Slingshot#
Slingshot 用于在低维空间中推断连续且可分支的 lineage 结构。它最初面向单细胞 RNA 测序数据中的发育轨迹建模,通常接在降维和聚类之后使用。它可以处理任意数量的分支事件,也允许通过半监督图构建的方式引入先验知识。
Traj=ov.single.TrajInfer(
adata,basis='X_umap',
groupby='clusters',
use_rep='scaled|original|X_pca',
n_comps=50
)
Traj.set_origin_cells('Ductal')
#Traj.set_terminal_cells(["Granule mature","OL","Astrocytes"])
如果只需要建议的伪时间排序,而暂时不需要检查 lineage 过程,可以不设置 debug_axes 参数。
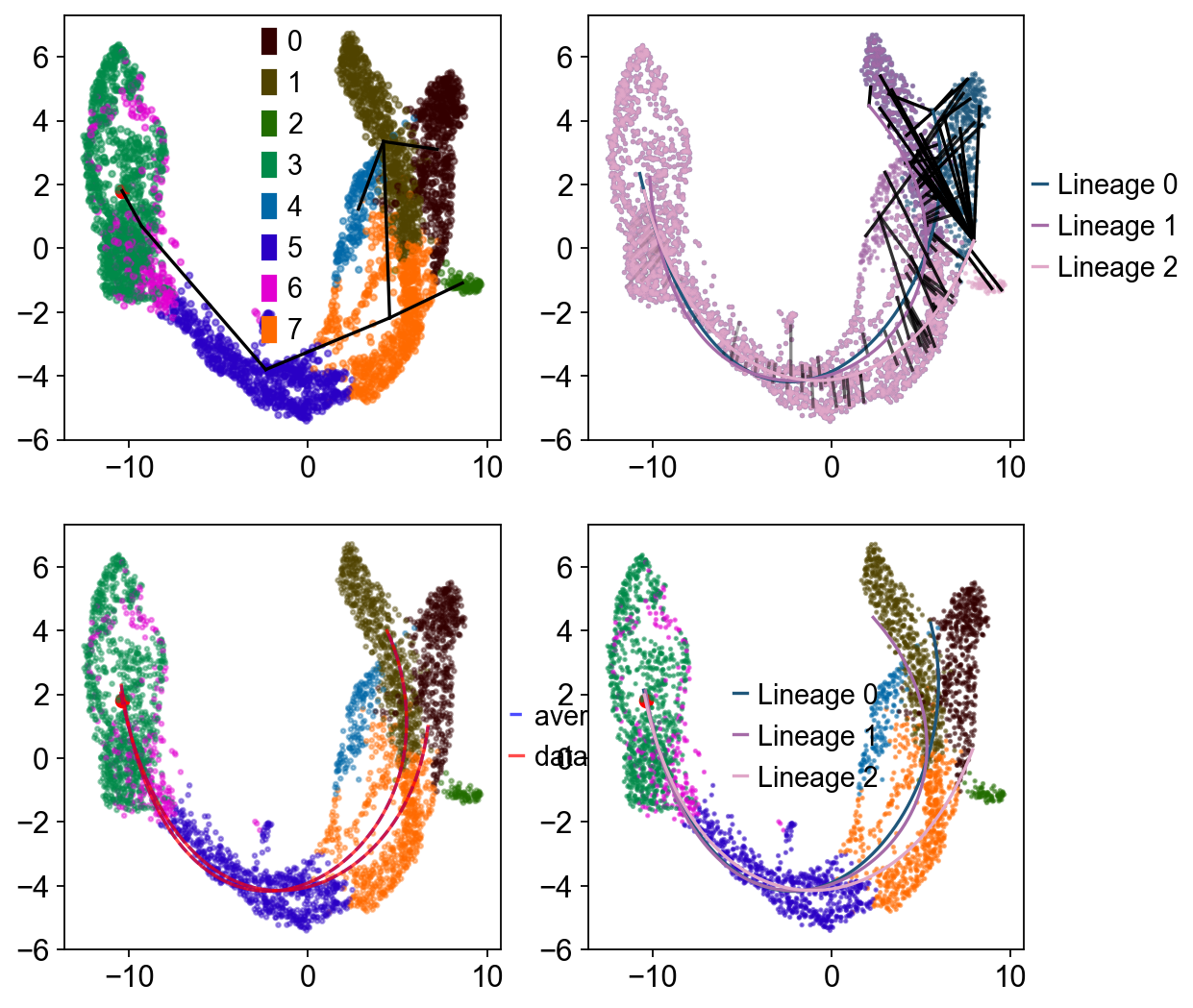
Traj.inference(method='slingshot',num_epochs=1)
Lineages: [Lineage[3, 6, 5, 7, 1, 0], Lineage[3, 6, 5, 7, 1, 4], Lineage[3, 6, 5, 7, 2]]
Reversing from leaf to root
Averaging branch @1 with lineages: [0, 1] [<pcurvepy2.pcurve.PrincipalCurve object at 0x15095efd0>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1517f46d0>]
Averaging branch @7 with lineages: [0, 1, 2] [<pcurvepy2.pcurve.PrincipalCurve object at 0x150c8dd90>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1510c62d0>]
Shrinking branch @7 with curves: [<pcurvepy2.pcurve.PrincipalCurve object at 0x150c8dd90>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1510c62d0>]
Shrinking branch @1 with curves: [<pcurvepy2.pcurve.PrincipalCurve object at 0x15095efd0>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1517f46d0>]
如果希望可视化 lineage 拟合过程,可以设置 debug_axes。
fig, axes = plt.subplots(nrows=2, ncols=2, figsize=(8, 8))
Traj.inference(method='slingshot',num_epochs=1,debug_axes=axes)
Lineages: [Lineage[3, 6, 5, 7, 1, 0], Lineage[3, 6, 5, 7, 1, 4], Lineage[3, 6, 5, 7, 2]]
Reversing from leaf to root
Averaging branch @1 with lineages: [0, 1] [<pcurvepy2.pcurve.PrincipalCurve object at 0x151880510>, <pcurvepy2.pcurve.PrincipalCurve object at 0x151133ed0>]
Averaging branch @7 with lineages: [0, 1, 2] [<pcurvepy2.pcurve.PrincipalCurve object at 0x151731e50>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1518bc8d0>]
Shrinking branch @7 with curves: [<pcurvepy2.pcurve.PrincipalCurve object at 0x151731e50>, <pcurvepy2.pcurve.PrincipalCurve object at 0x1518bc8d0>]
Shrinking branch @1 with curves: [<pcurvepy2.pcurve.PrincipalCurve object at 0x151880510>, <pcurvepy2.pcurve.PrincipalCurve object at 0x151133ed0>]
ov.pl.embedding(
adata,basis='X_umap',
color=['clusters','slingshot_pseudotime'],
frameon='small',
cmap='Reds'
)
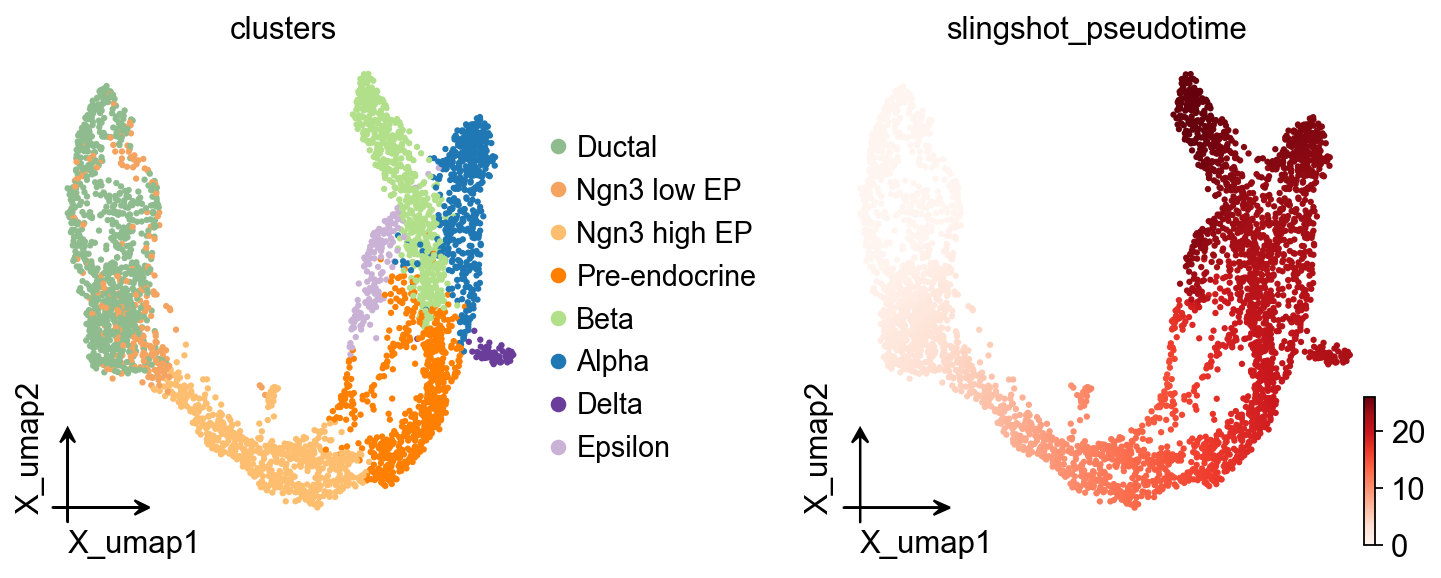
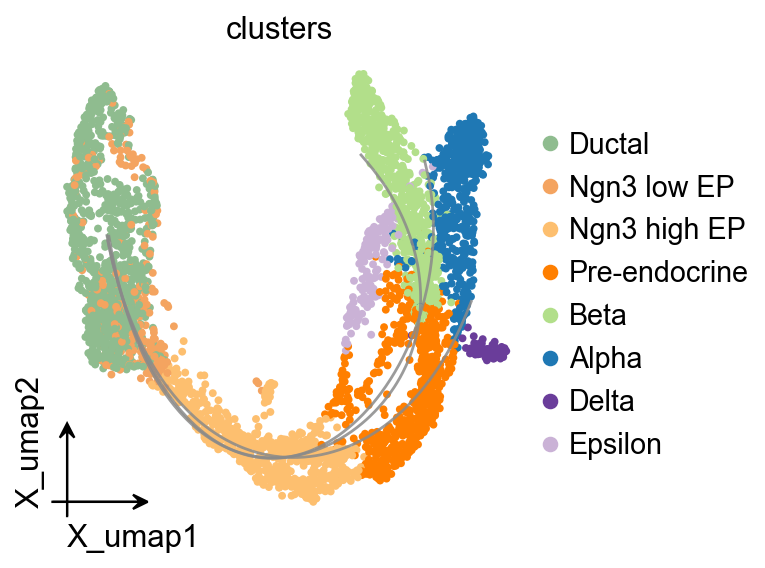
OV Slingshot 曲线叠加#
Slingshot 会在模型对象中保存拟合的谱系曲线,因此可以将其叠加到统一的 OmicVerse embedding 风格上。
fig, ax = plt.subplots(figsize=(4, 4))
ov.pl.embedding(
adata,
basis='X_umap',
color='clusters',
ax=ax,
show=False,
size=50,
)
ov.pl.trajectory_overlay(
adata,
ax=ax,
method='slingshot',
model=Traj.slingshot,
)
plt.show()
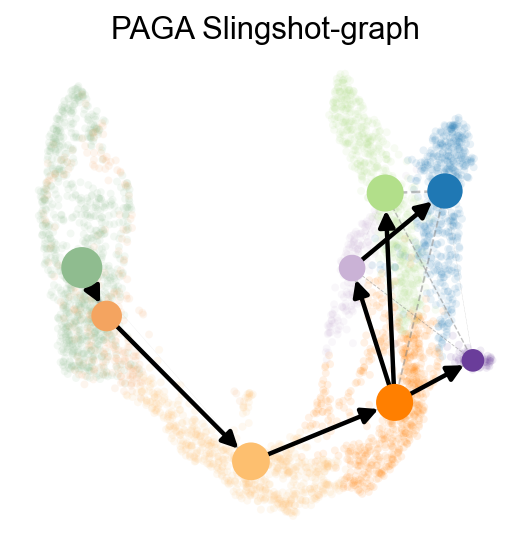
sc.pp.neighbors(adata,use_rep='scaled|original|X_pca')
ov.utils.cal_paga(
adata,
use_time_prior='slingshot_pseudotime',
vkey='paga',
groups='clusters'
)
running PAGA using priors: ['slingshot_pseudotime']
finished
added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns)
'paga/transitions_confidence', velocity transitions (adata.uns)
ov.utils.plot_paga(
adata,basis='umap',
size=50,
alpha=.1,
title='PAGA Slingshot-graph',
min_edge_width=2,
node_size_scale=1.5,
show=False,
legend_loc=False
)
<Axes: title={'center': 'PAGA Slingshot-graph'}>
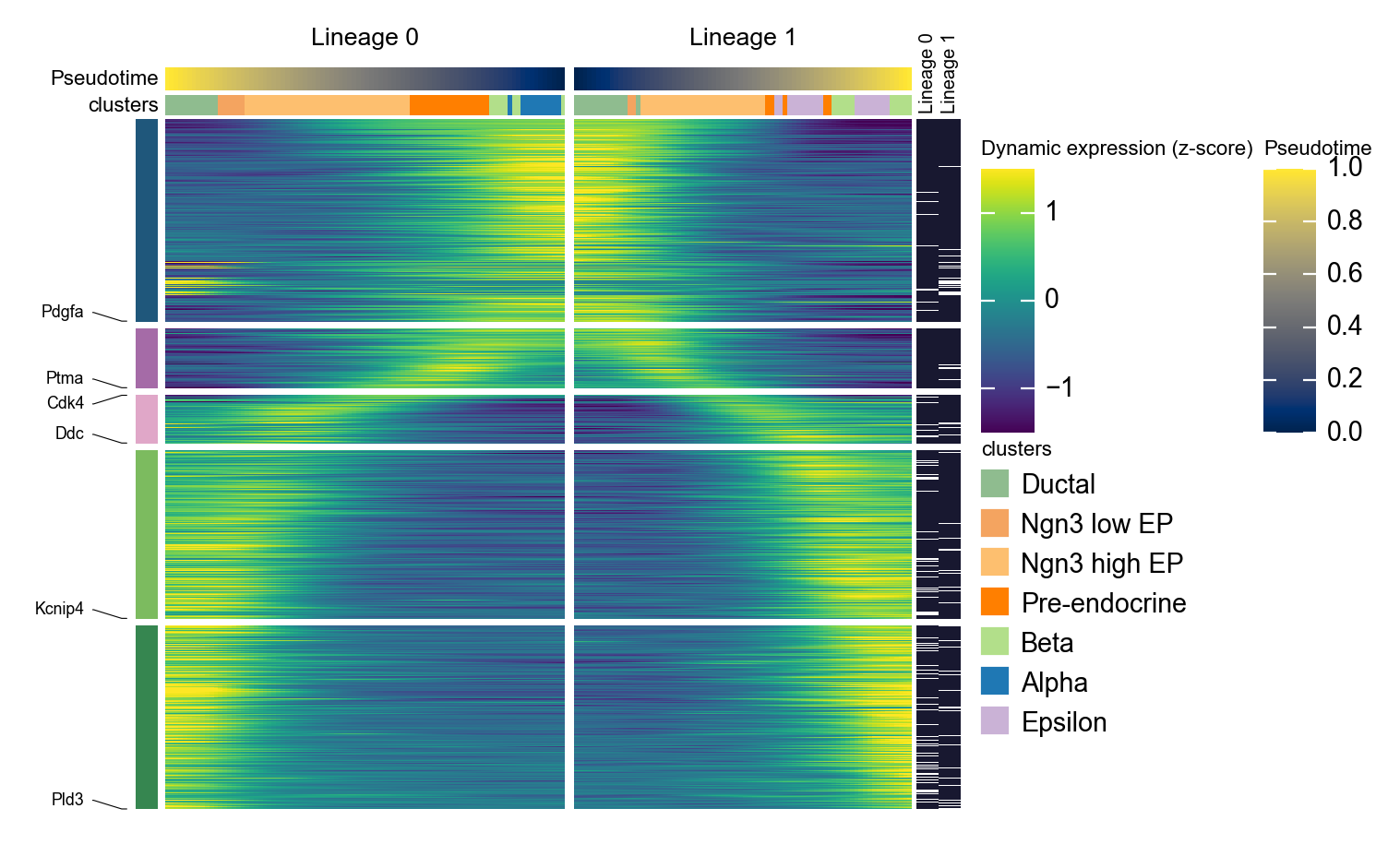
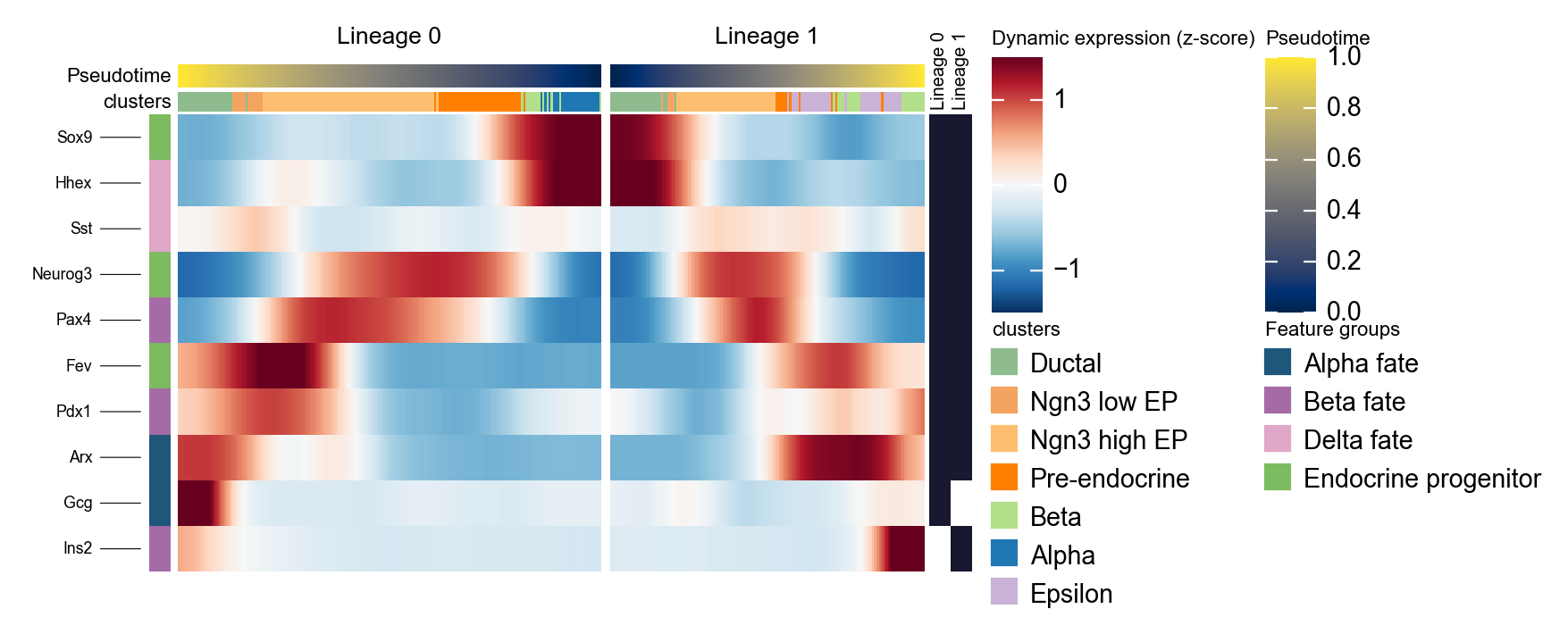
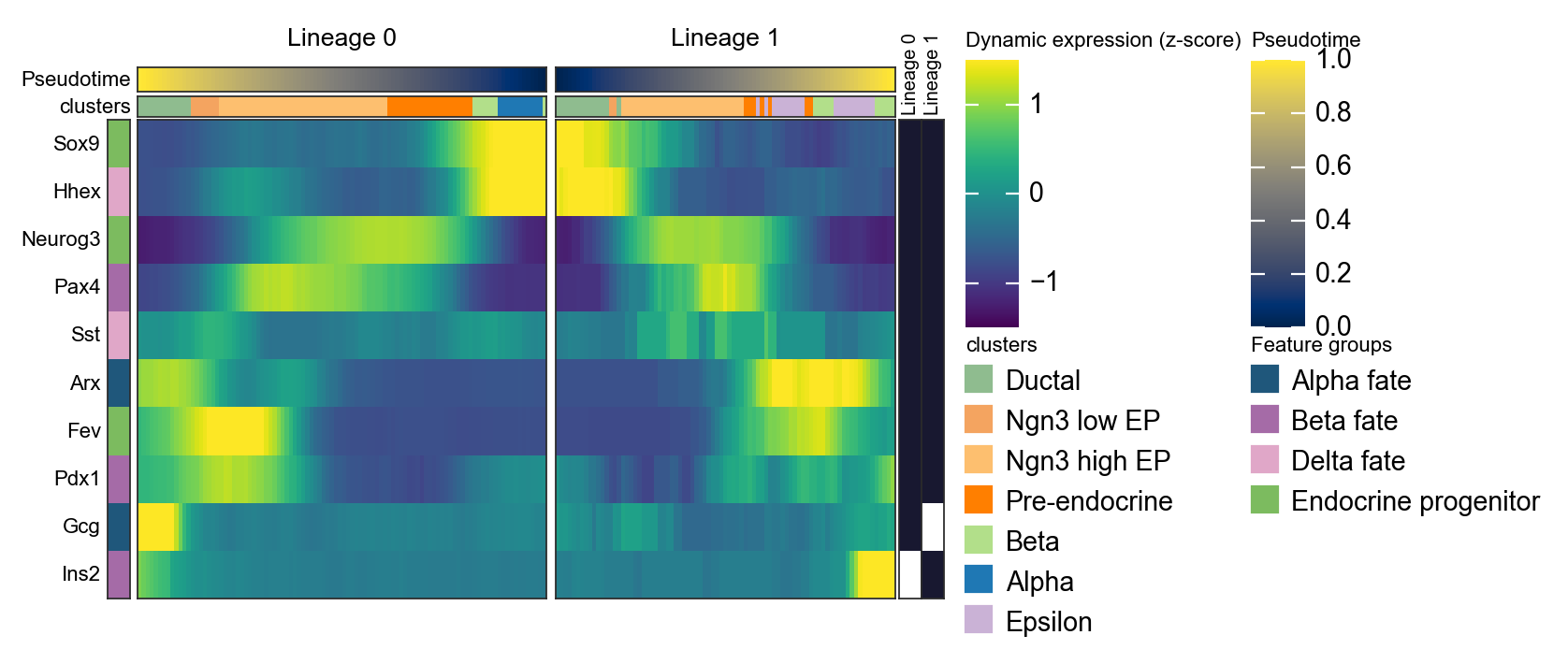
用镜像动态热图概括两条 Slingshot lineage#
Slingshot 可以返回多条 lineage 曲线。下面的代码会把拟合曲线转换成每个细胞的 lineage 标签和 lineage-specific pseudotime,然后并排绘制两条 lineage。这里反转第一条 lineage,使左侧面板指向分支点,这样更容易比较分支特异性程序。
import pandas as pd
import numpy as np
slingshot_genes = ['Sox9', 'Neurog3', 'Fev', 'Gcg', 'Arx', 'Pax4', 'Ins2', 'Pdx1', 'Sst', 'Hhex']
n_lineages = len(Traj.slingshot.lineages)
slingshot_lineage_labels = [f'Lineage {i}' for i in range(n_lineages)]
dominant_lineage = np.asarray(Traj.slingshot.cell_weights).argmax(axis=1)
lineage_specific_pt = np.full(adata.n_obs, np.nan)
for i, curve in enumerate(Traj.slingshot.curves):
curve_pt = np.asarray(curve.pseudotimes_interp, dtype=float)
adata.obs[f'slingshot_lineage_{i + 1}_pt'] = curve_pt
lineage_specific_pt[dominant_lineage == i] = curve_pt[dominant_lineage == i]
adata.obs['slingshot_lineage'] = pd.Categorical(
[slingshot_lineage_labels[i] for i in dominant_lineage],
categories=slingshot_lineage_labels,
ordered=True,
)
adata.obs['slingshot_lineage_pseudotime'] = lineage_specific_pt
selected_slingshot_lineages = slingshot_lineage_labels[:2]
slingshot_marker = {
'Alpha fate': ['Gcg', 'Arx'],
'Beta fate': ['Pax4', 'Ins2', 'Pdx1'],
'Delta fate': ['Sst', 'Hhex'],
'Endocrine progenitor': ['Sox9', 'Neurog3', 'Fev'],
}
d1 = ov.pl.dynamic_heatmap(
adata,
var_names=slingshot_marker,
pseudotime='slingshot_lineage_pseudotime',
lineage_key='slingshot_lineage',
lineages=selected_slingshot_lineages,
reverse_ht=[selected_slingshot_lineages[0]],
use_raw=adata.raw is not None,
use_cell_columns=False,
cell_annotation='clusters',
cell_bins=200,
smooth_window=17,
fitted_window=31,
figsize=(5, 4),
standard_scale='var',
cmap='RdBu_r',
use_fitted=True,
border=False,
show=False,
)
🔍 Dynamic heatmap:
Candidate features: 10
Pseudotime: slingshot_lineage_pseudotime
Lineage key: slingshot_lineage
Cell annotation: clusters
use_fitted=True | cell_bins=200 | cmap=RdBu_r
Lineages: Lineage 0, Lineage 1
✅ Dynamic heatmap completed!
✓ Matrix shape: 10 features × 337 columns
d1 = ov.pl.dynamic_heatmap(
adata,
var_names=slingshot_marker,
pseudotime='slingshot_lineage_pseudotime',
lineage_key='slingshot_lineage',
lineages=selected_slingshot_lineages,
reverse_ht=[selected_slingshot_lineages[0]],
use_raw=adata.raw is not None,
use_cell_columns=False,
cell_annotation='clusters',
figsize=(5, 4),
standard_scale='var',
show_row_names=True,
use_fitted=False,
border=True,
show=False,
)
🔍 Dynamic heatmap:
Candidate features: 10
Pseudotime: slingshot_lineage_pseudotime
Lineage key: slingshot_lineage
Cell annotation: clusters
use_fitted=False | cell_bins=100 | cmap=viridis
Lineages: Lineage 0, Lineage 1
✅ Dynamic heatmap completed!
✓ Matrix shape: 10 features × 183 columns
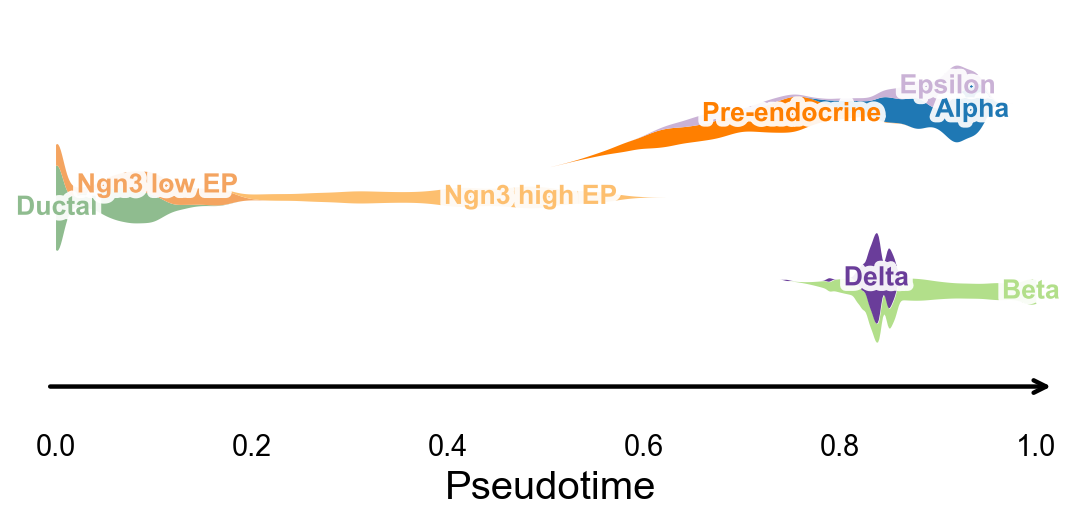
分支感知的伪时间流图#
ov.pl.branch_streamplot 只需要伪时间和细胞状态标签,因此也可以用于这个轨迹推断方法。图中 ribbon 的宽度表示某类细胞在伪时间上的富集位置,分叉的中心线则帮助观察不同 endocrine fate 在何处展开。
fig, ax = ov.pl.branch_streamplot(
adata,
group_key='clusters',
pseudotime_key='slingshot_pseudotime',
show=False,
)
plt.show()
Embedding stream plot#
在 Slingshot lineage 上使用 dynamic_features / dynamic_trends#
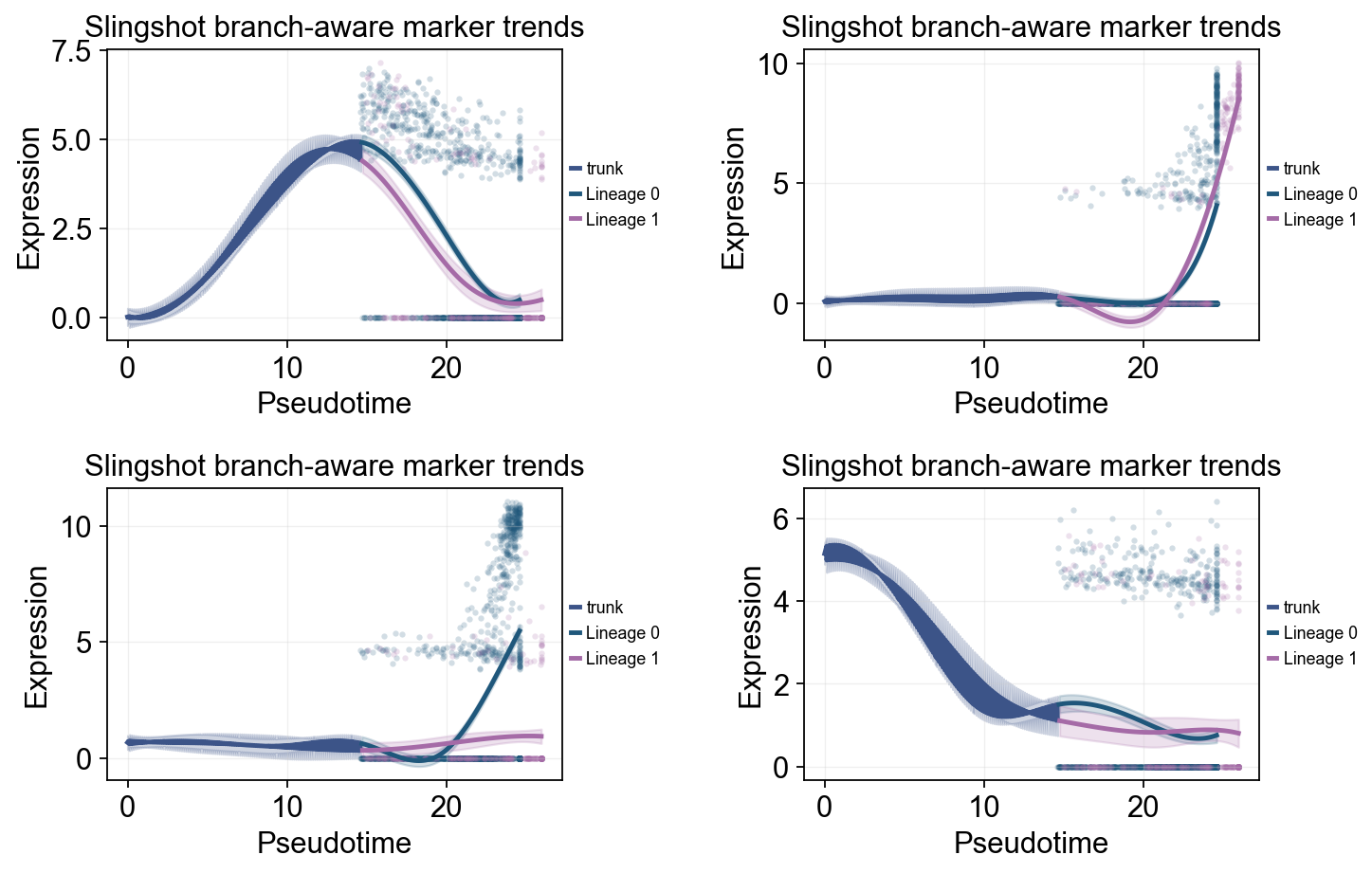
这里我们展示两种互补视角。第一种是全局趋势图:沿 slingshot_pseudotime 拟合 marker 曲线,并用 cluster 给原始散点着色。第二种是分支趋势图:按 Slingshot lineage 重新拟合,并在 endocrine split 之后比较各 lineage 的差异。
slingshot_trend_genes = ['Sox9', 'Neurog3', 'Fev', 'Gcg', 'Arx', 'Pax4', 'Ins2', 'Pdx1', 'Sst', 'Hhex']
slingshot_global_dyn = ov.single.dynamic_features(
adata,
genes=slingshot_trend_genes,
pseudotime='slingshot_pseudotime',
use_raw=adata.raw is not None,
distribution='normal',
link='identity',
n_splines=8,
store_raw=True,
raw_obs_keys=['clusters'],
)
🔍 Dynamic feature analysis:
Views: 1 | Features: 10
Pseudotime: slingshot_pseudotime
Stored raw obs keys: ['clusters']
Expression source: adata.raw
GAM: normal-identity | splines=8
✅ Dynamic feature analysis completed!
✓ Successful fits: 10/10
✓ Fitted rows: 2000
✓ Raw observations stored: 36960
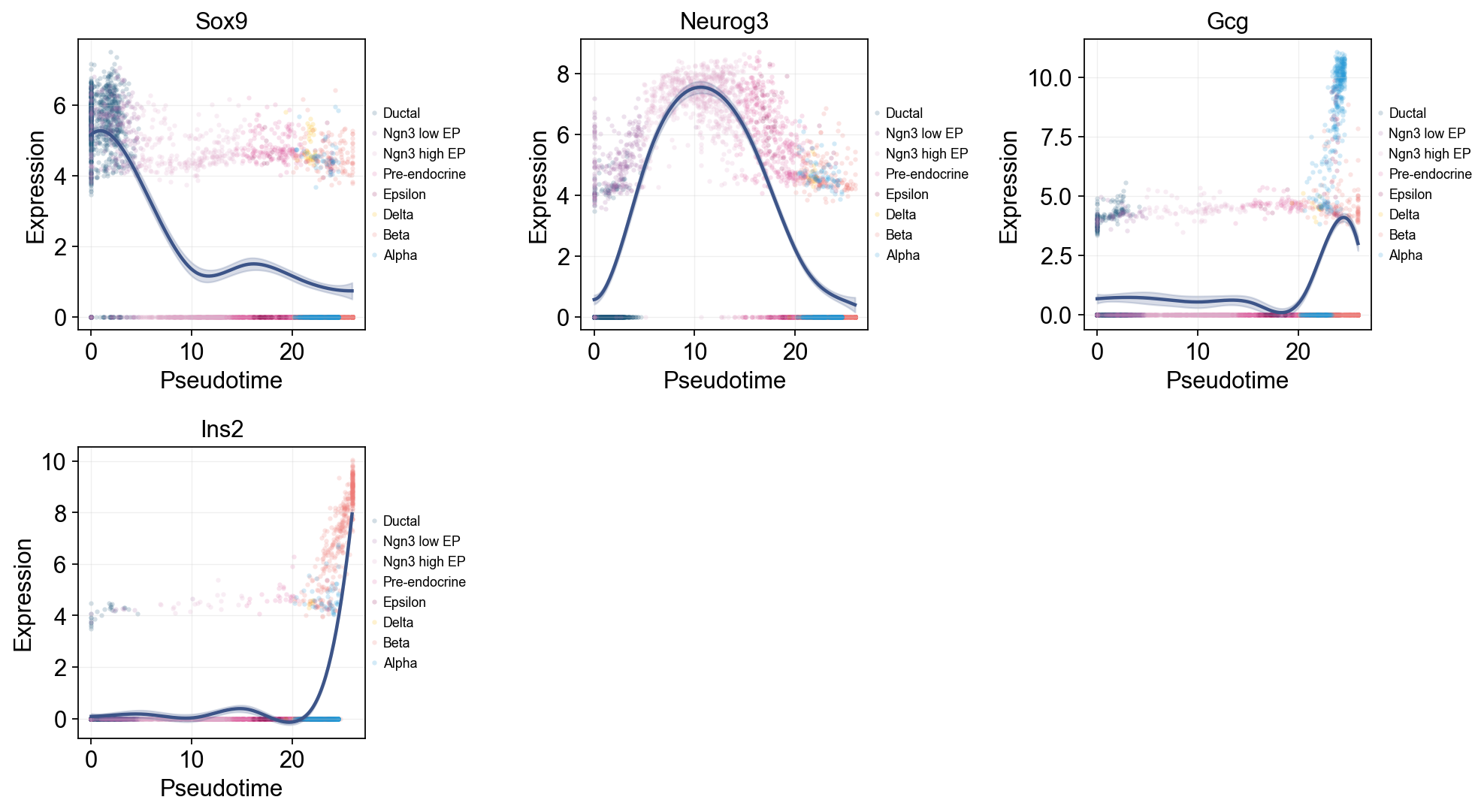
Single-line global trends#
这张图为每个基因只拟合一条全局趋势线,同时用细胞注释给原始散点着色。它适合区分“整体伪时间表达趋势”和“哪些细胞状态贡献了这些散点”。
ov.pl.dynamic_trends(
slingshot_global_dyn,
genes=['Sox9', 'Neurog3', 'Gcg', 'Ins2'],
add_point=True,
point_color_by='clusters',
figsize=(5, 3.5),
legend_loc='right margin',
legend_fontsize=8,
)
plt.show()
🔍 Dynamic trend plotting:
Features: 4 | Groups: 1
compare_features=False | compare_groups=False
✅ Dynamic trend plotting completed!
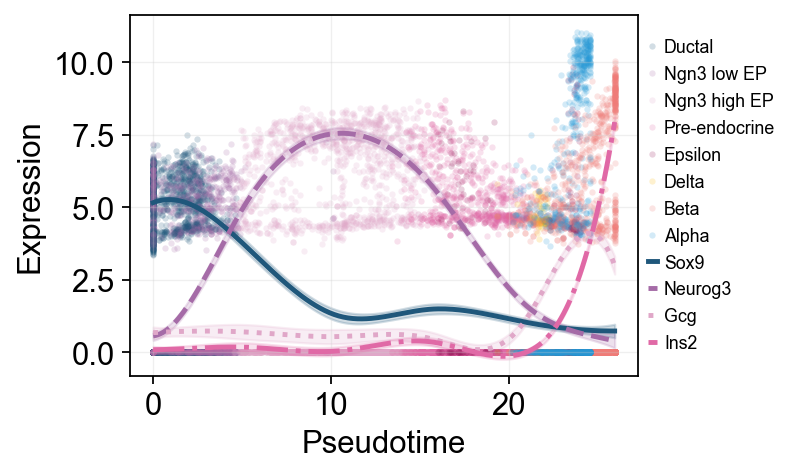
多标记基因趋势比较#
这里把多个 marker 的拟合曲线叠加在同一伪时间坐标中,方便直接比较不同程序的启动和衰减顺序。
ov.pl.dynamic_trends(
slingshot_global_dyn,
genes=['Sox9', 'Neurog3', 'Gcg', 'Ins2'],
compare_features=True,
add_point=True,
point_color_by='clusters',
line_style_by='features',
figsize=(6.2, 3.2),
linewidth=2.2,
legend_loc='right margin',
legend_fontsize=8,
)
plt.show()
🔍 Dynamic trend plotting:
Features: 4 | Groups: 1
compare_features=True | compare_groups=False
✅ Dynamic trend plotting completed!
slingshot_dyn_res = ov.single.dynamic_features(
adata,
genes=slingshot_trend_genes,
pseudotime='slingshot_lineage_pseudotime',
groupby='slingshot_lineage',
groups=selected_slingshot_lineages,
use_raw=adata.raw is not None,
distribution='normal',
link='identity',
n_splines=8,
store_raw=True,
)
slingshot_dyn_res.get_stats(successful_only=True).sort_values(['gene']).head(8)
🔍 Dynamic feature analysis:
Views: 2 | Features: 10
Pseudotime: slingshot_lineage_pseudotime
Grouping: slingshot_lineage
Expression source: adata.raw
GAM: normal-identity | splines=8
✅ Dynamic feature analysis completed!
✓ Successful fits: 20/20
✓ Fitted rows: 4000
✓ Raw observations stored: 30550
dataset groupby_key group gene success error n_cells \
14 Lineage 1 slingshot_lineage Lineage 1 Arx True None 533
4 Lineage 0 slingshot_lineage Lineage 0 Arx True None 2522
2 Lineage 0 slingshot_lineage Lineage 0 Fev True None 2522
12 Lineage 1 slingshot_lineage Lineage 1 Fev True None 533
3 Lineage 0 slingshot_lineage Lineage 0 Gcg True None 2522
13 Lineage 1 slingshot_lineage Lineage 1 Gcg True None 533
9 Lineage 0 slingshot_lineage Lineage 0 Hhex True None 2522
19 Lineage 1 slingshot_lineage Lineage 1 Hhex True None 533
exp_ncells peak_time valley_time min_pseudotime max_pseudotime \
14 110 20.163205 8.026130 0.0 25.970730
4 600 22.561005 7.726371 0.0 24.600767
2 1010 19.594078 9.086213 0.0 24.600767
12 182 20.032699 7.634611 0.0 25.970730
3 643 24.538956 18.234237 0.0 24.600767
13 83 25.252946 14.420933 0.0 25.970730
9 765 1.421652 24.291712 0.0 24.600767
19 188 2.283858 25.905477 0.0 25.970730
r2 explained_deviance p_value padj
14 0.395091 0.395091 7.481768e-01 7.481768e-01
4 0.265808 0.265808 1.936936e-01 1.936936e-01
2 0.589726 0.589726 1.110223e-16 1.387779e-16
12 0.406811 0.406811 1.110223e-16 1.586033e-16
3 0.318281 0.318281 1.110223e-16 1.387779e-16
13 0.016039 0.016039 1.160965e-07 1.451207e-07
9 0.488404 0.488404 1.110223e-16 1.387779e-16
19 0.563442 0.563442 1.110223e-16 1.586033e-16
slingshot_split_mask = adata.obs['clusters'].astype(str).isin(['Ngn3 high EP', 'Pre-endocrine'])
slingshot_split_time = float(np.nanmedian(adata.obs.loc[slingshot_split_mask, 'slingshot_lineage_pseudotime'])) if slingshot_split_mask.any() else float(np.nanmedian(adata.obs['slingshot_lineage_pseudotime']))
ov.pl.dynamic_trends(
slingshot_dyn_res,
genes=['Pax4', 'Ins2', 'Gcg', 'Sox9'],
compare_groups=True,
split_time=slingshot_split_time,
shared_trunk=True,
add_point=True,
point_color_by='group',
figsize=(5.5, 3),
linewidth=2.2,
ncols=2,
legend_loc='right margin',
legend_fontsize=8,
title='Slingshot branch-aware marker trends',
)
plt.show()
🔍 Dynamic trend plotting:
Features: 4 | Groups: 2
compare_features=False | compare_groups=True
✅ Dynamic trend plotting completed!
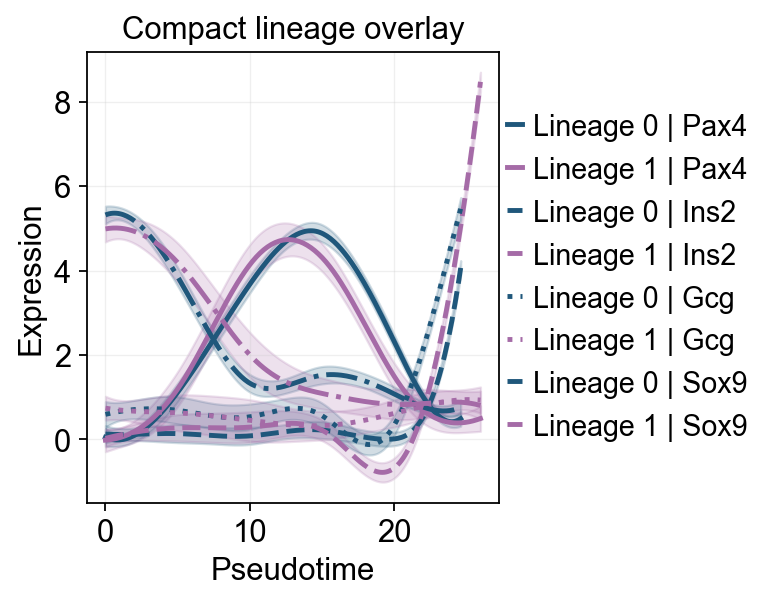
ov.pl.dynamic_trends(
slingshot_dyn_res,
genes=['Pax4', 'Ins2', 'Gcg', 'Sox9'],
compare_features=True,
compare_groups=True,
line_style_by='features',
figsize=(6, 4),
linewidth=2.2,
title='Compact lineage overlay',
)
plt.show()
🔍 Dynamic trend plotting:
Features: 4 | Groups: 2
compare_features=True | compare_groups=True
✅ Dynamic trend plotting completed!
g = ov.pl.dynamic_heatmap(
adata,
top_features=1000, # 保留动态性最强的前 1000 个基因用于绘图
pseudotime='slingshot_lineage_pseudotime',
lineage_key='slingshot_lineage',
lineages=selected_slingshot_lineages,
reverse_ht=[selected_slingshot_lineages[0]],
use_raw=adata.raw is not None,
use_cell_columns=False,
cell_annotation='clusters',
cell_bins=90,
smooth_window=17,
fitted_window=31,
n_split=5,
figsize=(5, 6),
standard_scale='var',
cmap='viridis',
top_label_features=10,
border=False,
show=False,
)
🔍 Dynamic heatmap:
Candidate features: 3000
Pseudotime: slingshot_lineage_pseudotime
Lineage key: slingshot_lineage
Cell annotation: clusters
use_fitted=True | cell_bins=90 | cmap=viridis
Lineages: Lineage 0, Lineage 1
✅ Dynamic heatmap completed!
✓ Matrix shape: 1000 features × 166 columns