使用kb-python进行Bulk RNA-seq比对#
本Notebook演示了在OmicVerse中使用kb-python(kallisto | bustools)的无比对bulk RNA-seq工作流程,从SRA文件到**差异表达(DE)**和可视化。
流程概述
导入OmicVerse并设置绘图风格
下载SRA数据(直接
.lite.1链接)将SRA转换为双端FASTQ(
parallel_fastq_dump)下载参考基因组/注释(Ensembl GRCh38)
构建kb参考(索引 + 转录本到基因映射)
使用kb-python定量reads(
ov.alignment.count,technology=BULK)将样本合并为单一矩阵,通过
ov.bulk.pyDEG运行DESeq2可视化DEG(火山图)
▲ 重要提示 本Notebook不修改任何现有代码或输出;仅在提供的单元格周围添加教程风格的Markdown说明。
步骤0 — 导入OmicVerse#
我们首先导入OmicVerse并为下游图形设置一致的绘图风格。
%%time
import omicverse as ov
ov.style(font_path='Arial')
🔬 Starting plot initialization...
Using already downloaded Arial font from: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] Tesla P40
Memory: 22.4 GB | Compute: 6.1
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.9rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
CPU times: user 7.88 s, sys: 2.53 s, total: 10.4 s
Wall time: 15.3 s
步骤1 — 下载SRA输入文件(直接.lite.1链接)#
这里我们下载几个示例SRA run。在此工作流程中,我们使用以.lite.1结尾的NCBI SRA直接下载链接。
▲ 重要提示
如果文件已存在,
ov.datasets.download_data(..., dir=...)将跳过下载。在许多环境中,您也可以通过
ov.alignment.prefetch()下载,然后使用fastq-dump/fasterq-dump进行转换。如果您的网络阻止了直接SRA链接,请切换到
prefetch或将文件镜像到可访问的位置。
links=[
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544419/SRR12544419.lite.1',#no
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-924/SRR012/12544/SRR12544421/SRR12544421.lite.1',#no
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544433/SRR12544433.lite.1'#yes
'https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos9/sra-pub-zq-922/SRR012/12544/SRR12544435/SRR12544435.lite.1'#yes
]
for link in links:
ov.datasets.download_data(link,dir='./data')
🔍 Downloading data to ./data/SRR12544433.lite.1
✅ Download completed
步骤2 — 将SRA转换为双端FASTQ(单个示例)#
此单元格展示了使用ov.alignment.parallel_fastq_dump将一个SRA文件转换为双端FASTQ。
▲ 重要提示
设置
split_files=True以获得双端输出(*_1.fastq.gz、*_2.fastq.gz)。将快速本地磁盘用于
tmpdir以减少I/O开销。线程数控制转换速度;根据CPU配置选择合适的值。
ov.alignment.parallel_fastq_dump(
sra_id='./data/SRR12544421.lite.1',
threads=12,
outdir='./data/SRR12544421',
tmpdir='./tmp',
split_files=True,
gzip=True,
)
🚀 Starting parallel-fastq-dump for ./data/SRR12544421.lite.1
Threads: 12
Output directory: ./data/SRR12544421
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544421.lite.1 --threads 12 --outdir ./data/SRR12544421 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:56:27,879 - SRR ids: ['./data/SRR12544421.lite.1']
2026-01-29 23:56:27,879 - extra args: ['--split-files', '--gzip']
2026-01-29 23:56:27,880 - tempdir: ./tmp/pfd_0armgw5l
2026-01-29 23:56:27,880 - CMD: sra-stat --meta --quick ./data/SRR12544421.lite.1
2026-01-29 23:56:27,982 - ./data/SRR12544421.lite.1 spots: 10963094
2026-01-29 23:56:27,982 - blocks: [[1, 913591], [913592, 1827182], [1827183, 2740773], [2740774, 3654364], [3654365, 4567955], [4567956, 5481546], [5481547, 6395137], [6395138, 7308728], [7308729, 8222319], [8222320, 9135910], [9135911, 10049501], [10049502, 10963094]]
2026-01-29 23:56:27,982 - CMD: fastq-dump -N 1 -X 913591 -O ./tmp/pfd_0armgw5l/0 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,985 - CMD: fastq-dump -N 913592 -X 1827182 -O ./tmp/pfd_0armgw5l/1 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,986 - CMD: fastq-dump -N 1827183 -X 2740773 -O ./tmp/pfd_0armgw5l/2 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,987 - CMD: fastq-dump -N 2740774 -X 3654364 -O ./tmp/pfd_0armgw5l/3 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,989 - CMD: fastq-dump -N 3654365 -X 4567955 -O ./tmp/pfd_0armgw5l/4 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,991 - CMD: fastq-dump -N 4567956 -X 5481546 -O ./tmp/pfd_0armgw5l/5 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,992 - CMD: fastq-dump -N 5481547 -X 6395137 -O ./tmp/pfd_0armgw5l/6 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,994 - CMD: fastq-dump -N 6395138 -X 7308728 -O ./tmp/pfd_0armgw5l/7 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,996 - CMD: fastq-dump -N 7308729 -X 8222319 -O ./tmp/pfd_0armgw5l/8 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:27,998 - CMD: fastq-dump -N 8222320 -X 9135910 -O ./tmp/pfd_0armgw5l/9 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:28,001 - CMD: fastq-dump -N 9135911 -X 10049501 -O ./tmp/pfd_0armgw5l/10 --split-files --gzip ./data/SRR12544421.lite.1
2026-01-29 23:56:28,003 - CMD: fastq-dump -N 10049502 -X 10963094 -O ./tmp/pfd_0armgw5l/11 --split-files --gzip ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913593 spots for ./data/SRR12544421.lite.1
Written 913593 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
Read 913591 spots for ./data/SRR12544421.lite.1
Written 913591 spots for ./data/SRR12544421.lite.1
✓ parallel-fastq-dump completed successfully!
{'sra_id': './data/SRR12544421.lite.1',
'threads': 12,
'outdir': './data/SRR12544421',
'split_files': True,
'gzip': True}
步骤2b — 批量转换多个SRA文件#
此循环使用相同设置将多个SRA文件转换为双端FASTQ。
提示:如果有大量样本,建议使用作业调度器(如Slurm)跨节点并行处理,而不是在单个节点上无限增加线程数。
for sra in [
'SRR12544433','SRR12544435'
]:
ov.alignment.parallel_fastq_dump(
sra_id=f'./data/{sra}.lite.1',
threads=12,
outdir=f'./data/{sra}',
tmpdir='./tmp',
split_files=True,
gzip=True,
)
🚀 Starting parallel-fastq-dump for ./data/SRR12544433.lite.1
Threads: 12
Output directory: ./data/SRR12544433
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544433.lite.1 --threads 12 --outdir ./data/SRR12544433 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:58:48,245 - SRR ids: ['./data/SRR12544433.lite.1']
2026-01-29 23:58:48,245 - extra args: ['--split-files', '--gzip']
2026-01-29 23:58:48,246 - tempdir: ./tmp/pfd_8tgtda45
2026-01-29 23:58:48,246 - CMD: sra-stat --meta --quick ./data/SRR12544433.lite.1
2026-01-29 23:58:48,358 - ./data/SRR12544433.lite.1 spots: 16602881
2026-01-29 23:58:48,358 - blocks: [[1, 1383573], [1383574, 2767146], [2767147, 4150719], [4150720, 5534292], [5534293, 6917865], [6917866, 8301438], [8301439, 9685011], [9685012, 11068584], [11068585, 12452157], [12452158, 13835730], [13835731, 15219303], [15219304, 16602881]]
2026-01-29 23:58:48,359 - CMD: fastq-dump -N 1 -X 1383573 -O ./tmp/pfd_8tgtda45/0 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,361 - CMD: fastq-dump -N 1383574 -X 2767146 -O ./tmp/pfd_8tgtda45/1 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,362 - CMD: fastq-dump -N 2767147 -X 4150719 -O ./tmp/pfd_8tgtda45/2 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,364 - CMD: fastq-dump -N 4150720 -X 5534292 -O ./tmp/pfd_8tgtda45/3 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,365 - CMD: fastq-dump -N 5534293 -X 6917865 -O ./tmp/pfd_8tgtda45/4 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,367 - CMD: fastq-dump -N 6917866 -X 8301438 -O ./tmp/pfd_8tgtda45/5 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,368 - CMD: fastq-dump -N 8301439 -X 9685011 -O ./tmp/pfd_8tgtda45/6 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,370 - CMD: fastq-dump -N 9685012 -X 11068584 -O ./tmp/pfd_8tgtda45/7 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,372 - CMD: fastq-dump -N 11068585 -X 12452157 -O ./tmp/pfd_8tgtda45/8 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,373 - CMD: fastq-dump -N 12452158 -X 13835730 -O ./tmp/pfd_8tgtda45/9 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,375 - CMD: fastq-dump -N 13835731 -X 15219303 -O ./tmp/pfd_8tgtda45/10 --split-files --gzip ./data/SRR12544433.lite.1
2026-01-29 23:58:48,377 - CMD: fastq-dump -N 15219304 -X 16602881 -O ./tmp/pfd_8tgtda45/11 --split-files --gzip ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383578 spots for ./data/SRR12544433.lite.1
Written 1383578 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
Read 1383573 spots for ./data/SRR12544433.lite.1
Written 1383573 spots for ./data/SRR12544433.lite.1
✓ parallel-fastq-dump completed successfully!
🚀 Starting parallel-fastq-dump for ./data/SRR12544435.lite.1
Threads: 12
Output directory: ./data/SRR12544435
Added to PATH: /home/groups/xiaojie/steorra/env/omicverse/bin
>> /home/groups/xiaojie/steorra/env/omicverse/bin/parallel-fastq-dump --sra-id ./data/SRR12544435.lite.1 --threads 12 --outdir ./data/SRR12544435 --tmpdir ./tmp --minSpotId 1 --split-files --gzip
2026-01-29 23:59:23,325 - SRR ids: ['./data/SRR12544435.lite.1']
2026-01-29 23:59:23,325 - extra args: ['--split-files', '--gzip']
2026-01-29 23:59:23,326 - tempdir: ./tmp/pfd_t7s3okn4
2026-01-29 23:59:23,326 - CMD: sra-stat --meta --quick ./data/SRR12544435.lite.1
2026-01-29 23:59:23,433 - ./data/SRR12544435.lite.1 spots: 16486139
2026-01-29 23:59:23,433 - blocks: [[1, 1373844], [1373845, 2747688], [2747689, 4121532], [4121533, 5495376], [5495377, 6869220], [6869221, 8243064], [8243065, 9616908], [9616909, 10990752], [10990753, 12364596], [12364597, 13738440], [13738441, 15112284], [15112285, 16486139]]
2026-01-29 23:59:23,433 - CMD: fastq-dump -N 1 -X 1373844 -O ./tmp/pfd_t7s3okn4/0 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,435 - CMD: fastq-dump -N 1373845 -X 2747688 -O ./tmp/pfd_t7s3okn4/1 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,436 - CMD: fastq-dump -N 2747689 -X 4121532 -O ./tmp/pfd_t7s3okn4/2 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,438 - CMD: fastq-dump -N 4121533 -X 5495376 -O ./tmp/pfd_t7s3okn4/3 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,439 - CMD: fastq-dump -N 5495377 -X 6869220 -O ./tmp/pfd_t7s3okn4/4 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,441 - CMD: fastq-dump -N 6869221 -X 8243064 -O ./tmp/pfd_t7s3okn4/5 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,443 - CMD: fastq-dump -N 8243065 -X 9616908 -O ./tmp/pfd_t7s3okn4/6 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,445 - CMD: fastq-dump -N 9616909 -X 10990752 -O ./tmp/pfd_t7s3okn4/7 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,451 - CMD: fastq-dump -N 10990753 -X 12364596 -O ./tmp/pfd_t7s3okn4/8 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,453 - CMD: fastq-dump -N 12364597 -X 13738440 -O ./tmp/pfd_t7s3okn4/9 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,455 - CMD: fastq-dump -N 13738441 -X 15112284 -O ./tmp/pfd_t7s3okn4/10 --split-files --gzip ./data/SRR12544435.lite.1
2026-01-29 23:59:23,456 - CMD: fastq-dump -N 15112285 -X 16486139 -O ./tmp/pfd_t7s3okn4/11 --split-files --gzip ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373855 spots for ./data/SRR12544435.lite.1
Written 1373855 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
Read 1373844 spots for ./data/SRR12544435.lite.1
Written 1373844 spots for ./data/SRR12544435.lite.1
✓ parallel-fastq-dump completed successfully!
步骤3 — 下载参考基因组 + 注释#
我们从Ensembl下载GRCh38参考FASTA和GTF。
▲ 重要提示
保持FASTA和GTF来自相同的Ensembl版本以避免转录本ID不匹配。
确保有足够的磁盘空间(人类参考文件较大)。
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-108/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz',
dir='./genomes')
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-108/gtf/homo_sapiens/Homo_sapiens.GRCh38.108.gtf.gz',
dir='./genomes')
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
✅ Download completed
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.108.gtf.gz
✅ Download completed
'./genomes/Homo_sapiens.GRCh38.108.gtf.gz'
步骤4 — 构建kb-python参考(索引 + t2g + cDNA)#
ov.alignment.single.ref(...)准备kb-python参考资源:
index.idx:kallisto索引t2g.txt:用于将计数聚合到基因的转录本到基因映射cdna.fa:从注释派生的转录组FASTA
▲ 重要提示
首次构建可能耗时较长;构建后可对所有样本复用相同的输出。
将索引放在快速存储设备上以加快定量速度。
result = ov.alignment.single.ref(
fasta_paths='genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz', #input
gtf_paths='genomes/Homo_sapiens.GRCh38.108.gtf.gz', #input
index_path='pbmc_1k_v3/index.idx', #output
t2g_path='pbmc_1k_v3/t2g.txt', #output
cdna_path='pbmc_1k_v3/cdna.fa', #output
temp_dir='tmp',
overwrite=True,
)
print(result.keys())
🚀 Starting ref workflow: standard
Using temporary directory: tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb ref --tmp tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -t 8 --overwrite --d-list-overhang 1 -f1 pbmc_1k_v3/cdna.fa pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz pbmc_1k_v3/Homo_sapiens.GRCh38.108.gtf.gz
[2026-01-29 22:21:41,856] INFO [ref] Preparing pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz, pbmc_1k_v3/Homo_sapiens.GRCh38.108.gtf.gz
[2026-01-29 22:22:33,317] INFO [ref] Splitting genome pbmc_1k_v3/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz into cDNA at /scratch/users/steorra/analysis/26_omic_protocol/tmp-kb-e9513f2c23aa46d1961cde1d7afeba6b/tmpoyd3o7ta
[2026-01-29 22:23:30,907] INFO [ref] Concatenating 1 cDNAs to pbmc_1k_v3/cdna.fa
[2026-01-29 22:23:31,551] INFO [ref] Creating transcript-to-gene mapping at pbmc_1k_v3/t2g.txt
[2026-01-29 22:23:33,230] INFO [ref] Indexing pbmc_1k_v3/cdna.fa to pbmc_1k_v3/index.idx
✓ ref workflow completed!
dict_keys(['workflow', 'technology', 'parameters', 'index_path', 't2g_path', 'cdna_path'])
Step 5 — Quantify one sample with kb-python (technology = BULK)#
This is an example call for one sample.
▲ CRITICAL
For bulk RNA-seq, each sample is treated as one “library” (no barcode filtering).
h5ad=Truewrites an AnnData object for convenient downstream analysis in OmicVerse/Scanpy.If your environment does not expose
count(...)directly, the equivalent OmicVerse entry point is typicallyov.alignment.count(...)(the rest of the arguments stay the same).
result = count(
fastq_paths=[
"./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz",
"./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz",
],
index_path="pbmc_1k_v3/index.idx",
t2g_path="pbmc_1k_v3/t2g.txt",
technology='BULK', # technology
output_path="results/pbmc_test",
h5ad=True,
filter_barcodes=False,
threads=12,
parity="paired", #
strand="unstranded", #
)
print(result.keys())
🚀 Starting count workflow: standard
Technology: BULK
Output directory: .
Using temporary directory: tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o . -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-29 22:53:46,113] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to . from
[2026-01-29 22:53:46,113] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz
[2026-01-29 22:53:46,113] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-29 22:54:23,070] INFO [count] Sorting BUS file ./output.bus to tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:25,476] INFO [count] Inspecting BUS file tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:26,580] INFO [count] Generating count matrix ./counts_unfiltered/cells_x_genes from BUS file tmp-kb-6e8cfea103a042e79f4e83e5bd1facf2/output.s.bus
[2026-01-29 22:54:28,602] INFO [count] Writing gene names to file ./counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-29 22:54:28,835] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-29 22:54:28,860] INFO [count] Reading matrix ./counts_unfiltered/cells_x_genes.mtx
[2026-01-29 22:54:28,917] INFO [count] Writing matrix to h5ad ./counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
步骤5b — 定量所有样本(循环)#
我们对每个样本运行kb-python定量,并将结果保存到每个样本的输出文件夹中。
提示:在实践中,建议为output_path设置清晰的命名规范(项目/样本/日期)以保持结果的可重现性。
for sra in [
'SRR12544419','SRR12544421','SRR12544433','SRR12544435'
]:
result = ov.alignment.count(
fastq_paths=[
f"./data/{sra}/{sra}.lite.1_1.fastq.gz",
f"./data/{sra}/{sra}.lite.1_2.fastq.gz",
],
index_path="pbmc_1k_v3/index.idx",
t2g_path="pbmc_1k_v3/t2g.txt",
technology='BULK', # technology
output_path=f"results/{sra}/",
h5ad=True,
filter_barcodes=False,
threads=12,
parity="paired", # ✅关键
strand="unstranded", # ✅建议显式写
)
print(result.keys())
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544419/
Using temporary directory: tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544419/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-30 00:03:00,630] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544419/ from
[2026-01-30 00:03:00,630] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_1.fastq.gz
[2026-01-30 00:03:00,630] INFO [count] ./data/SRR12544419/SRR12544419.lite.1_2.fastq.gz
[2026-01-30 00:03:35,180] INFO [count] Sorting BUS file results/SRR12544419/output.bus to tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:37,594] INFO [count] Inspecting BUS file tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:38,702] INFO [count] Generating count matrix results/SRR12544419/counts_unfiltered/cells_x_genes from BUS file tmp-kb-36a26e9c1a61444387dc9ef0c9f177dc/output.s.bus
[2026-01-30 00:03:41,325] INFO [count] Writing gene names to file results/SRR12544419/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:03:41,560] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:03:41,586] INFO [count] Reading matrix results/SRR12544419/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:03:41,643] INFO [count] Writing matrix to h5ad results/SRR12544419/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544421/
Using temporary directory: tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544421/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544421/SRR12544421.lite.1_1.fastq.gz ./data/SRR12544421/SRR12544421.lite.1_2.fastq.gz
[2026-01-30 00:03:53,957] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544421/ from
[2026-01-30 00:03:53,957] INFO [count] ./data/SRR12544421/SRR12544421.lite.1_1.fastq.gz
[2026-01-30 00:03:53,957] INFO [count] ./data/SRR12544421/SRR12544421.lite.1_2.fastq.gz
[2026-01-30 00:04:18,493] INFO [count] Sorting BUS file results/SRR12544421/output.bus to tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:20,598] INFO [count] Inspecting BUS file tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:21,702] INFO [count] Generating count matrix results/SRR12544421/counts_unfiltered/cells_x_genes from BUS file tmp-kb-7dc3eb1f1a7f46369c65439c1df20d7e/output.s.bus
[2026-01-30 00:04:23,724] INFO [count] Writing gene names to file results/SRR12544421/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:04:23,956] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:04:23,981] INFO [count] Reading matrix results/SRR12544421/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:04:24,037] INFO [count] Writing matrix to h5ad results/SRR12544421/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544433/
Using temporary directory: tmp-kb-f36592f2f361482c8829daecfd59d0a4
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-f36592f2f361482c8829daecfd59d0a4 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544433/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544433/SRR12544433.lite.1_1.fastq.gz ./data/SRR12544433/SRR12544433.lite.1_2.fastq.gz
[2026-01-30 00:04:33,656] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544433/ from
[2026-01-30 00:04:33,656] INFO [count] ./data/SRR12544433/SRR12544433.lite.1_1.fastq.gz
[2026-01-30 00:04:33,656] INFO [count] ./data/SRR12544433/SRR12544433.lite.1_2.fastq.gz
[2026-01-30 00:05:04,901] INFO [count] Sorting BUS file results/SRR12544433/output.bus to tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:07,209] INFO [count] Inspecting BUS file tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:08,313] INFO [count] Generating count matrix results/SRR12544433/counts_unfiltered/cells_x_genes from BUS file tmp-kb-f36592f2f361482c8829daecfd59d0a4/output.s.bus
[2026-01-30 00:05:10,436] INFO [count] Writing gene names to file results/SRR12544433/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:05:10,667] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:05:10,693] INFO [count] Reading matrix results/SRR12544433/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:05:10,749] INFO [count] Writing matrix to h5ad results/SRR12544433/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
🚀 Starting count workflow: standard
Technology: BULK
Output directory: results/SRR12544435/
Using temporary directory: tmp-kb-7e11e84640b14489ad063057ba8b2738
>> /home/groups/xiaojie/steorra/env/omicverse/bin/kb count --tmp tmp-kb-7e11e84640b14489ad063057ba8b2738 -i pbmc_1k_v3/index.idx -g pbmc_1k_v3/t2g.txt -x BULK -o results/SRR12544435/ -t 12 -m 2G --h5ad --parity paired --strand unstranded ./data/SRR12544435/SRR12544435.lite.1_1.fastq.gz ./data/SRR12544435/SRR12544435.lite.1_2.fastq.gz
[2026-01-30 00:05:21,328] INFO [count] Using index pbmc_1k_v3/index.idx to generate BUS file to results/SRR12544435/ from
[2026-01-30 00:05:21,328] INFO [count] ./data/SRR12544435/SRR12544435.lite.1_1.fastq.gz
[2026-01-30 00:05:21,328] INFO [count] ./data/SRR12544435/SRR12544435.lite.1_2.fastq.gz
[2026-01-30 00:05:52,473] INFO [count] Sorting BUS file results/SRR12544435/output.bus to tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:54,882] INFO [count] Inspecting BUS file tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:55,986] INFO [count] Generating count matrix results/SRR12544435/counts_unfiltered/cells_x_genes from BUS file tmp-kb-7e11e84640b14489ad063057ba8b2738/output.s.bus
[2026-01-30 00:05:58,108] INFO [count] Writing gene names to file results/SRR12544435/counts_unfiltered/cells_x_genes.genes.names.txt
[2026-01-30 00:05:58,341] WARNING [count] 22736 gene IDs do not have corresponding valid gene names. These genes will use their gene IDs instead.
[2026-01-30 00:05:58,366] INFO [count] Reading matrix results/SRR12544435/counts_unfiltered/cells_x_genes.mtx
[2026-01-30 00:05:58,424] INFO [count] Writing matrix to h5ad results/SRR12544435/counts_unfiltered/adata.h5ad
✓ count workflow completed!
dict_keys(['workflow', 'technology', 'output_path', 'parameters'])
步骤6 — 加载每个样本的结果并统一基因标识符#
每次kb-python运行都会生成一个adata.h5ad文件及配套的基因名称文件。
我们加载每个样本的AnnData对象,并确保adata.var同时包含:
gene_namegene_id
▲ 重要提示
不同流程中的基因命名规范可能不同;明确设置adata.var['gene_name']并将其用作索引,可使下游合并和可视化更加稳健。
ad_dict={}
for sra in [
'SRR12544419','SRR12544421','SRR12544433','SRR12544435'
]:
ad=ov.read(f'./results/{sra}/counts_unfiltered/adata.h5ad')
gene_name=ov.pd.read_csv(
f'./results/{sra}/counts_unfiltered/cells_x_genes.genes.names.txt',
header=None
)
ad.var['gene_name']=gene_name[0].tolist()
ad.var['gene_id']=ad.var.index
ad.var.index=ad.var['gene_name']
ad.var_names_make_unique()
ad.obs['sra']=sra
ad_dict[sra]=ad
Step 7 — Merge samples and define phenotype labels#
We concatenate all samples into one AnnData and create a Group column (e.g., disease vs healthy) for downstream DE.
▲ CRITICAL
For bulk RNA-seq, you typically want one observation per sample. In this demo workflow, each sample’s output is concatenated and then labeled via adata.obs['Group'].
adata=ov.concat(ad_dict)
adata.obs_names_make_unique()
adata.obs['Group']=['no','no','yes','yes']
adata
AnnData object with n_obs × n_vars = 4 × 62703
obs: 'sra', 'Group'
步骤7b — 快速合理性检查(基因表达)#
对标记基因进行快速检查,有助于确认矩阵已正确加载,并在预期位置包含非零计数。
adata[:,'CD3D'].X.toarray()
array([[623.],
[612.],
[ 98.],
[324.]])
步骤8 — 转换为DE分析用的计数矩阵#
ov.bulk.pyDEG期望一个基因 × 样本计数矩阵(pandas DataFrame)。
这里我们将AnnData转换为DataFrame并进行转置以符合该约定。
▲ 重要提示 DESeq2风格的方法需要原始整数计数。不要在DESeq2之前应用对数归一化。
data=adata.to_df().T
data.head()
| barcode | AAAAAAAAAAAAAAAA | AAAAAAAAAAAAAAAA-1 | AAAAAAAAAAAAAAAA-2 | AAAAAAAAAAAAAAAA-3 |
|---|---|---|---|---|
| gene_name | ||||
| ATAD3B | 90.0 | 70.0 | 30.0 | 115.0 |
| DDX11L17 | 4.0 | 0.0 | 0.0 | 0.0 |
| ENSG00000228037.1 | 9.0 | 8.0 | 1.0 | 8.0 |
| PRDM16 | 0.0 | 0.0 | 0.0 | 0.0 |
| ENSG00000284616.1 | 0.0 | 0.0 | 0.0 | 0.0 |
步骤9 — 创建pyDEG对象#
我们将计数矩阵封装到OmicVerse的pyDEG对象中,用于差异表达分析。
dds=ov.bulk.pyDEG(data)
步骤10 — 差异表达(DESeq2)#
我们使用DESeq2后端运行DE分析。
▲ 重要提示
DESeq2期望原始整数计数,并在内部估算大小因子/离散度。
这里的
treatment_groups和control_groups指的是提供矩阵中的列/样本标识符。在您自己的数据中,请替换为适当的样本ID(通常是样本名称)。
dds.drop_duplicates_index()
print('... drop_duplicates_index success')
treatment_groups=['AAAAAAAAAAAAAAAA-2','AAAAAAAAAAAAAAAA-3']
control_groups=['AAAAAAAAAAAAAAAA','AAAAAAAAAAAAAAAA-1']
result=dds.deg_analysis(treatment_groups,control_groups,method='DEseq2')
... drop_duplicates_index success
⚙️ You are using DEseq2 method for differential expression analysis.
⏰ Start to create DeseqDataSet...
logres_prior=1.2473932639674563, sigma_prior=0.25
Log2 fold change & Wald test p-value: condition Treatment vs Control
baseMean log2FoldChange lfcSE stat \
gene_name
B2M 178491.812733 0.065096 0.418607 0.155506
S100A9 170557.796728 0.378402 0.248436 1.523133
S100A8 154015.888583 0.914504 0.455013 2.009840
MT-RNR2 143254.671256 0.434606 0.553576 0.785089
HBB 127090.703257 2.661912 1.449787 1.836071
... ... ... ... ...
ENSG00000273937.1 0.000000 NaN NaN NaN
ENSG00000278633.1 0.000000 NaN NaN NaN
ENSG00000278066.1 0.000000 NaN NaN NaN
ENSG00000277374.1 0.000000 NaN NaN NaN
ENSG00000275661.1 0.000000 NaN NaN NaN
pvalue padj
gene_name
B2M 0.876422 0.996897
S100A9 0.127725 0.669610
S100A8 0.044448 0.402458
MT-RNR2 0.432402 0.948016
HBB 0.066347 0.493618
... ... ...
ENSG00000273937.1 NaN NaN
ENSG00000278633.1 NaN NaN
ENSG00000278066.1 NaN NaN
ENSG00000277374.1 NaN NaN
ENSG00000275661.1 NaN NaN
[62703 rows x 6 columns]
✅ Differential expression analysis completed.
步骤11 — 过滤低表达基因#
过滤可以去除信号不足的基因,提高可解释性和多重检验行为。
这里我们在计算DE后应用BaseMean阈值。
print(result.shape)
result=result.loc[result['log2(BaseMean)']>1]
print(result.shape)
(21002, 14)
(21002, 14)
步骤12 — 设置DEG阈值#
我们设置显著性和倍数变化的阈值,这些阈值控制DEG的判定和下游可视化。
# -1 means automatically calculates
dds.foldchange_set(fc_threshold=-1,
pval_threshold=0.05,
logp_max=10)
... Fold change threshold: 2.9757964388302165
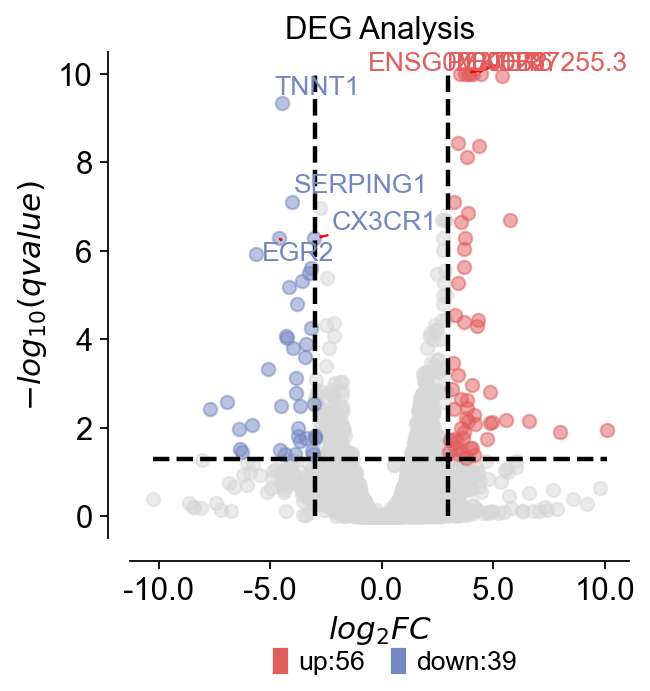
步骤13 — 可视化DEG(火山图)#
火山图汇总了效应大小(log2FC)与显著性之间的关系。
提示:识别DEG后,典型的后续步骤包括:
基因集富集分析(GO/KEGG/Reactome)
通路可视化
PPI网络分析
(根据您的教程范围,可以将这些下游步骤作为附加部分添加。)
dds.plot_volcano(title='DEG Analysis',figsize=(4,4),
plot_genes_num=8,plot_genes_fontsize=12,)
🌋 Volcano Plot Analysis:
Total genes: 62703
↗️ Upregulated genes: 56
↘️ Downregulated genes: 39
➡️ Non-significant genes: 62608
🎯 Total significant genes: 95
log2FC range: -10.26 to 10.10
qvalue range: 5.94e-14 to 1.00e+00
⚙️ Current Function Parameters:
Data columns: pval_name='qvalue', fc_name='log2FC'
Thresholds: pval_threshold=0.05, fc_max=2.9757964388302165, fc_min=-2.9757964388302165
Plot size: figsize=(4, 4)
Gene labels: plot_genes_num=8, plot_genes_fontsize=12
Custom genes: None (auto-select top genes)
💡 Parameter Optimization Suggestions:
✅ Current parameters are optimal for your data!
────────────────────────────────────────────────────────────
<Axes: title={'center': 'DEG Analysis'}, xlabel='$log_{2}FC$', ylabel='$-log_{10}(qvalue)$'>
扩展性与可重现性注意事项#
CPU和内存: kb-python通常比全基因组比对更轻量,但索引构建和定量仍受益于多线程和快速磁盘。
参考一致性: 始终保持FASTA/GTF版本一致(并在教程中记录版本信息)。
项目结构: 考虑将输出组织为
results/<project>/<sample>/...,并保存描述组/重复的metadata.csv。
如有需要,我可以按照相同的OmicVerse教程风格,通过添加新的Markdown + 新的代码单元格来扩展本Notebook,加入富集分析和PPI分析内容(不触及现有单元格)。