使用STAR进行Bulk RNA-seq比对#
本Notebook演示了OmicVerse中端到端的bulk RNA-seq工作流程,从原始reads到差异表达(DE)结果和可视化。
工作流程
导入OmicVerse并设置绘图风格
从SRA下载双端FASTQ
质量控制和修剪(fastp)
下载参考基因组和注释(Ensembl GRCh38 release 115)
使用STAR比对reads
基因水平定量(featureCounts)
差异表达和可视化(pyDEG)
注意
完整教程系列可以从DE继续进行功能富集和PPI网络分析。本Notebook专注于核心预处理 + DE工作流程。
依赖要求
外部工具:
fasterq-dump(SRA Toolkit)、fastp、STAR和featureCounts(Subread)。存储:FASTQ + 比对输出文件可能很大;请确保有足够的磁盘空间。
内存:STAR基因组索引/比对可能需要大量内存(见下方STAR步骤)。
1) 导入OmicVerse#
我们首先导入OmicVerse并配置一致的绘图风格。如果本地没有该字体,ov.style(font_path='Arial')将自动下载/安装。
import omicverse as ov
ov.style(font_path='Arial')
🔬 Starting plot initialization...
Downloading Arial font from GitHub...
Arial font downloaded successfully to: /tmp/omicverse_arial.ttf
Registered as: Arial
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 1.7.9rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
2) 从SRA下载FASTQ数据#
我们下载4个双端样本作为轻量示例:
疾病组:
SRR12544419、SRR12544421健康组:
SRR12544529、SRR12544531
▲ 重要提示
ov.alignment.fqdump()将优先使用在sra_dir中找到的预下载SRA文件。如果未找到,将根据library_layout下载数据并转换为FASTQ格式。
fq_data = ov.alignment.fqdump(
# Download 4 samples as example
sra_ids=['SRR12544419','SRR12544421',#disease group
'SRR12544529','SRR12544531',# healthy group
],
sra_dir = "./data",
temp_dir = './data/tmp',
output_dir="./data/fqdata",
library_layout="paired", # pair-end data
)
print(fq_data)
>> /home/groups/xiaojie/steorra/env/omicverse/bin/fasterq-dump SRR12544529 -O data/fqdata/SRR12544529 -e 8 --mem 4G --split-files --progress -t data/tmp/SRR12544529
>> /home/groups/xiaojie/steorra/env/omicverse/bin/fasterq-dump SRR12544421 -O data/fqdata/SRR12544421 -e 8 --mem 4G --split-files --progress -t data/tmp/SRR12544421
>> /home/groups/xiaojie/steorra/env/omicverse/bin/fasterq-dump SRR12544531 -O data/fqdata/SRR12544531 -e 8 --mem 4G --split-files --progress -t data/tmp/SRR12544531
>> /home/groups/xiaojie/steorra/env/omicverse/bin/fasterq-dump SRR12544419 -O data/fqdata/SRR12544419 -e 8 --mem 4G --split-files --progress -t data/tmp/SRR12544419
join :| 0.00% 0.01% 0.02% 0.03% 0.04% 0.05% 0.06% 0.07% 0.08% 0.09% 0.10% 0.11% 0.12% 0.13% 0.14% 0.15% 0.16% 0.17% 0.18% 0.19% 0.20% 0.21% 0.22% 0.23% 0.24% 0.25% 0.26% 0.27% 0.28% 0.29% 0.30% 0.31% 0.32% 0.33% 0.34% 0.35% 0.36% 0.37% 0.38% 0.39% 0.40% 0.41% 0.42% 0.43% 0.44% 0.45% 0.46% 0.47% 0.48% 0.49% 0.50% 0.51% 0.52% 0.53% 0.54% 0.55% 0.56% 0.57% 0.58% 0.59% 0.60% 0.61% 0.62% 0.63% 0.64% 0.65% 0.66% 0.67% 0.68% 0.69% 0.70% 0.71% 0.72% 0.73% 0.74% 0.75% 0.76% 0.77% 0.78% 0.79% 0.80% 0.81% 0.82% 0.83% 0.84% 0.85% 0.86% 0.87% 0.88% 0.89% 0.90% 0.91% 0.92% 0.93% 0.94% 0.95% 0.96% 0.97% 0.98% 0.99%- 1.00% 1.01% 1.02% 1.03% 1.04% 1.05% 1.06% 1.07% 1.08% 1.09% 1.10% 1.11% 1.12% 1.13% 1.14% 1.15% 1.16% 1.17% 1.18% 1.19% 1.20% 1.21% 1.22% 1.23% 1.24% 1.25% 1.26% 1.27% 1.28% 1.29% 1.30% 1.31% 1.32% 1.33% 1.34% 1.35% 1.36% 1.37% 1.38% 1.39% 1.40% 1.41% 1.42% 1.43% 1.44% 1.45% 1.46% 1.47% 1.48% 1.49% 1.50% 1.51% 1.52% 1.53% 1.54% 1.55% 1.56% 1.57% 1.58% 1.59% 1.60% 1.61% 1.62% 1.63% 1.64% 1.65% 1.66% 1.67% 1.68% 1.69% 1.70% 1.71% 1.72% 1.73% 1.74% 1.75% 1.76% 1.77% 1.78% 1.79% 1.80% 1.81% 1.82% 1.83% 1.84% 1.85% 1.86% 1.87% 1.88% 1.89% 1.90% 1.91% 1.92% 1.93% 1.94% 1.95% 1.96% 1.97% 1.98% 1.99% 2.00% 2.01% 2.02% 2.03% 2.04% 2.05% 2.06% 2.07% 2.08% 2.09% 2.10% 2.11% 2.12% 2.13% 2.14% 2.15% 2.16% 2.17% 2.18% 2.19% 2.20% 2.21% 2.22% 2.23% 2.24% 2.25% 2.26% 2.27% 2.28% 2.29% 2.30% 2.31% 2.32% 2.33% 2.34% 2.35% 2.36% 2.37% 2.38% 2.39% 2.40% 2.41% 2.42% 2.43% 2.44% 2.45% 2.46% 2.47% 2.48% 2.49% 2.50% 2.51% 2.52% 2.53% 2.54% 2.55% 2.56% 2.57% 2.58% 2.59% 2.60% 2.61% 2.62% 2.63% 2.64% 2.65% 2.66% 2.67% 2.68% 2.69% 2.70% 2.71% 2.72% 2.73% 2.74% 2.75% 2.76% 2.77% 2.78% ------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 10,963,094
reads read : 21,926,188
reads written : 21,926,188
join :|-------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 16,415,795
reads read : 32,831,590
reads written : 32,831,590
join :|-------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 18,850,866
reads read : 37,701,732
reads written : 37,701,732
join :|-------------------------------------------------- 100%
concat :|-------------------------------------------------- 100%
spots read : 20,038,226
reads read : 40,076,452
reads written : 40,076,452
[{'srr': 'SRR12544419', 'fq1': 'data/fqdata/SRR12544419/SRR12544419_1.fastq', 'fq2': 'data/fqdata/SRR12544419/SRR12544419_2.fastq', 'layout': 'paired'}, {'srr': 'SRR12544421', 'fq1': 'data/fqdata/SRR12544421/SRR12544421_1.fastq', 'fq2': 'data/fqdata/SRR12544421/SRR12544421_2.fastq', 'layout': 'paired'}, {'srr': 'SRR12544529', 'fq1': 'data/fqdata/SRR12544529/SRR12544529_1.fastq', 'fq2': 'data/fqdata/SRR12544529/SRR12544529_2.fastq', 'layout': 'paired'}, {'srr': 'SRR12544531', 'fq1': 'data/fqdata/SRR12544531/SRR12544531_1.fastq', 'fq2': 'data/fqdata/SRR12544531/SRR12544531_2.fastq', 'layout': 'paired'}]
3) 质量控制和修剪(fastp)#
我们对每个样本运行fastp并保存清洗后的FASTQ文件及QC报告。
输出文件
清洗后的reads:
./result/fqdata/<sample>/*_clean_*.fastq报告:
*.fastp.json和*.fastp.html
⏱️ 运行时间:对于这个小型示例,通常需要数十秒,具体取决于计算和磁盘性能。
from operator import itemgetter
qc_result = ov.alignment.fastp(
samples=list(map(itemgetter("srr", "fq1", "fq2"), fq_data)),
output_dir = './result/fqdata')
print(qc_result)
>> /home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544421/SRR12544421_1.fastq -o result/fqdata/SRR12544421/SRR12544421_clean_1.fastq -w 8 -j result/fqdata/SRR12544421/SRR12544421.fastp.json -h result/fqdata/SRR12544421/SRR12544421.fastp.html -I data/fqdata/SRR12544421/SRR12544421_2.fastq -O result/fqdata/SRR12544421/SRR12544421_clean_2.fastq --detect_adapter_for_pe
>> /home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544419/SRR12544419_1.fastq -o result/fqdata/SRR12544419/SRR12544419_clean_1.fastq -w 8 -j result/fqdata/SRR12544419/SRR12544419.fastp.json -h result/fqdata/SRR12544419/SRR12544419.fastp.html -I data/fqdata/SRR12544419/SRR12544419_2.fastq -O result/fqdata/SRR12544419/SRR12544419_clean_2.fastq --detect_adapter_for_pe
>> /home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544531/SRR12544531_1.fastq -o result/fqdata/SRR12544531/SRR12544531_clean_1.fastq -w 8 -j result/fqdata/SRR12544531/SRR12544531.fastp.json -h result/fqdata/SRR12544531/SRR12544531.fastp.html -I data/fqdata/SRR12544531/SRR12544531_2.fastq -O result/fqdata/SRR12544531/SRR12544531_clean_2.fastq --detect_adapter_for_pe
>> /home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544529/SRR12544529_1.fastq -o result/fqdata/SRR12544529/SRR12544529_clean_1.fastq -w 8 -j result/fqdata/SRR12544529/SRR12544529.fastp.json -h result/fqdata/SRR12544529/SRR12544529.fastp.html -I data/fqdata/SRR12544529/SRR12544529_2.fastq -O result/fqdata/SRR12544529/SRR12544529_clean_2.fastq --detect_adapter_for_pe
Detecting adapter sequence for read1...
Detecting adapter sequence for read1...
Detecting adapter sequence for read1...
Detecting adapter sequence for read1...
CTCCTGGAAGTTAACTGCACCATCAGTGTTGATATCCAACTCTTTGAACCAGACGTCTGC
Detecting adapter sequence for read2...
CTGGAAGTTAACTGCACCATCAGTGTTGATATCCAACTCTTTGAACCAGACGTCTGCACC
Detecting adapter sequence for read2...
No adapter detected for read1
Detecting adapter sequence for read2...
GGCCACTGTGGTCTTAGGGGGTGCCCTCCCCGAGGCCTGGCTTATGGTGGTGGCCAGGGC
Detecting adapter sequence for read2...
GTGGCTCCTCGGCTTTGACAGAGTGCAAGACGATGACTTGCAAAATGTCGCAGCTGGAAC
GTGGCTCCTCGGCTTTGACAGAGTGCAAGACGATGACTTGCAAAATGTCGCAGCTGGAAC
GACACAACTGTGTTCACTAGCAACCTCAAACAGACACCATGGTGCACCTGACTCCTGAGG
GTGGCTCCTCGGCTTTGACAGAGTGCAAGACGATGACTTGCAAAATGTCGCAGCTGGAAC
Read1 before filtering:
total reads: 10963094
total bases: 559117794
Q20 bases: 553775494(99.0445%)
Q30 bases: 541507863(96.8504%)
Q40 bases: 0(0%)
Read2 before filtering:
total reads: 10963094
total bases: 559117794
Q20 bases: 544098436(97.3137%)
Q30 bases: 526334946(94.1367%)
Q40 bases: 0(0%)
Read1 after filtering:
total reads: 10687072
total bases: 544418300
Q20 bases: 539356482(99.0702%)
Q30 bases: 527511135(96.8945%)
Q40 bases: 0(0%)
Read2 after filtering:
total reads: 10687072
total bases: 544746875
Q20 bases: 537103434(98.5969%)
Q30 bases: 521266217(95.6896%)
Q40 bases: 0(0%)
Filtering result:
reads passed filter: 21374144
reads failed due to low quality: 487504
reads failed due to too many N: 1192
reads failed due to too short: 63348
reads failed due to adapter dimer: 0
reads with adapter trimmed: 134829
bases trimmed due to adapters: 2669503
Duplication rate: 15.5483%
Insert size peak (evaluated by paired-end reads): 71
JSON report: result/fqdata/SRR12544421/SRR12544421.fastp.json
HTML report: result/fqdata/SRR12544421/SRR12544421.fastp.html
/home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544421/SRR12544421_1.fastq -o result/fqdata/SRR12544421/SRR12544421_clean_1.fastq -w 8 -j result/fqdata/SRR12544421/SRR12544421.fastp.json -h result/fqdata/SRR12544421/SRR12544421.fastp.html -I data/fqdata/SRR12544421/SRR12544421_2.fastq -O result/fqdata/SRR12544421/SRR12544421_clean_2.fastq --detect_adapter_for_pe
fastp v1.1.0, time used: 84 seconds
Read1 before filtering:
total reads: 20038226
total bases: 1021949526
Q20 bases: 1012475638(99.073%)
Q30 bases: 990432190(96.916%)
Q40 bases: 0(0%)
Read2 before filtering:
total reads: 20038226
total bases: 1021949526
Q20 bases: 1003879697(98.2318%)
Q30 bases: 975667178(95.4712%)
Q40 bases: 0(0%)
Read1 after filtering:
total reads: 19745691
total bases: 1006911636
Q20 bases: 997775728(99.0927%)
Q30 bases: 976169789(96.9469%)
Q40 bases: 0(0%)
Read2 after filtering:
total reads: 19745691
total bases: 1006642443
Q20 bases: 994858257(98.8294%)
Q30 bases: 968362375(96.1973%)
Q40 bases: 0(0%)
Filtering result:
reads passed filter: 39491382
reads failed due to low quality: 415354
reads failed due to too many N: 2198
reads failed due to too short: 167518
reads failed due to adapter dimer: 0
reads with adapter trimmed: 146355
bases trimmed due to adapters: 5132033
Duplication rate: 23.3277%
Insert size peak (evaluated by paired-end reads): 70
JSON report: result/fqdata/SRR12544529/SRR12544529.fastp.json
HTML report: result/fqdata/SRR12544529/SRR12544529.fastp.html
/home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544529/SRR12544529_1.fastq -o result/fqdata/SRR12544529/SRR12544529_clean_1.fastq -w 8 -j result/fqdata/SRR12544529/SRR12544529.fastp.json -h result/fqdata/SRR12544529/SRR12544529.fastp.html -I data/fqdata/SRR12544529/SRR12544529_2.fastq -O result/fqdata/SRR12544529/SRR12544529_clean_2.fastq --detect_adapter_for_pe
fastp v1.1.0, time used: 117 seconds
Read1 before filtering:
total reads: 18850866
total bases: 961394166
Q20 bases: 952217279(99.0455%)
Q30 bases: 931109128(96.8499%)
Q40 bases: 0(0%)
Read2 before filtering:
total reads: 18850866
total bases: 961394166
Q20 bases: 931425457(96.8828%)
Q30 bases: 899828659(93.5962%)
Q40 bases: 0(0%)
Read1 after filtering:
total reads: 18263658
total bases: 930838151
Q20 bases: 922174625(99.0693%)
Q30 bases: 901889848(96.8901%)
Q40 bases: 0(0%)
Read2 after filtering:
total reads: 18263658
total bases: 930927790
Q20 bases: 917031645(98.5073%)
Q30 bases: 889662854(95.5673%)
Q40 bases: 0(0%)
Filtering result:
reads passed filter: 36527316
reads failed due to low quality: 1068046
reads failed due to too many N: 2060
reads failed due to too short: 103592
reads failed due to adapter dimer: 718
reads with adapter trimmed: 209233
bases trimmed due to adapters: 4077359
Duplication rate: 17.3068%
Insert size peak (evaluated by paired-end reads): 71
JSON report: result/fqdata/SRR12544419/SRR12544419.fastp.json
HTML report: result/fqdata/SRR12544419/SRR12544419.fastp.html
/home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544419/SRR12544419_1.fastq -o result/fqdata/SRR12544419/SRR12544419_clean_1.fastq -w 8 -j result/fqdata/SRR12544419/SRR12544419.fastp.json -h result/fqdata/SRR12544419/SRR12544419.fastp.html -I data/fqdata/SRR12544419/SRR12544419_2.fastq -O result/fqdata/SRR12544419/SRR12544419_clean_2.fastq --detect_adapter_for_pe
fastp v1.1.0, time used: 125 seconds
Read1 before filtering:
total reads: 16415795
total bases: 837205545
Q20 bases: 829026406(99.023%)
Q30 bases: 810217511(96.7764%)
Q40 bases: 0(0%)
Read2 before filtering:
total reads: 16415795
total bases: 837205545
Q20 bases: 818343741(97.7471%)
Q30 bases: 794204257(94.8637%)
Q40 bases: 0(0%)
Read1 after filtering:
total reads: 15957703
total bases: 812314973
Q20 bases: 804542876(99.0432%)
Q30 bases: 786432142(96.8137%)
Q40 bases: 0(0%)
Read2 after filtering:
total reads: 15957703
total bases: 813371311
Q20 bases: 803301599(98.762%)
Q30 bases: 781597078(96.0935%)
Q40 bases: 0(0%)
Filtering result:
reads passed filter: 31915406
reads failed due to low quality: 576316
reads failed due to too many N: 1802
reads failed due to too short: 338066
reads failed due to adapter dimer: 0
reads with adapter trimmed: 365804
bases trimmed due to adapters: 11104213
Duplication rate: 36.3719%
Insert size peak (evaluated by paired-end reads): 51
JSON report: result/fqdata/SRR12544531/SRR12544531.fastp.json
HTML report: result/fqdata/SRR12544531/SRR12544531.fastp.html
/home/users/steorra/miniforge3/bin/fastp -i data/fqdata/SRR12544531/SRR12544531_1.fastq -o result/fqdata/SRR12544531/SRR12544531_clean_1.fastq -w 8 -j result/fqdata/SRR12544531/SRR12544531.fastp.json -h result/fqdata/SRR12544531/SRR12544531.fastp.html -I data/fqdata/SRR12544531/SRR12544531_2.fastq -O result/fqdata/SRR12544531/SRR12544531_clean_2.fastq --detect_adapter_for_pe
fastp v1.1.0, time used: 141 seconds
[{'sample': 'SRR12544419', 'clean1': 'result/fqdata/SRR12544419/SRR12544419_clean_1.fastq', 'clean2': 'result/fqdata/SRR12544419/SRR12544419_clean_2.fastq', 'json': 'result/fqdata/SRR12544419/SRR12544419.fastp.json', 'html': 'result/fqdata/SRR12544419/SRR12544419.fastp.html'}, {'sample': 'SRR12544421', 'clean1': 'result/fqdata/SRR12544421/SRR12544421_clean_1.fastq', 'clean2': 'result/fqdata/SRR12544421/SRR12544421_clean_2.fastq', 'json': 'result/fqdata/SRR12544421/SRR12544421.fastp.json', 'html': 'result/fqdata/SRR12544421/SRR12544421.fastp.html'}, {'sample': 'SRR12544529', 'clean1': 'result/fqdata/SRR12544529/SRR12544529_clean_1.fastq', 'clean2': 'result/fqdata/SRR12544529/SRR12544529_clean_2.fastq', 'json': 'result/fqdata/SRR12544529/SRR12544529.fastp.json', 'html': 'result/fqdata/SRR12544529/SRR12544529.fastp.html'}, {'sample': 'SRR12544531', 'clean1': 'result/fqdata/SRR12544531/SRR12544531_clean_1.fastq', 'clean2': 'result/fqdata/SRR12544531/SRR12544531_clean_2.fastq', 'json': 'result/fqdata/SRR12544531/SRR12544531.fastp.json', 'html': 'result/fqdata/SRR12544531/SRR12544531.fastp.html'}]
4) 下载参考基因组和基因注释(Ensembl)#
我们下载:
GRCh38主要组装FASTA
GRCh38 GTF注释(release 115)
提示:将这些文件缓存到本地以避免重复下载。
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-115/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz',dir='./genomes')
ov.datasets.download_data('ftp://ftp.ensembl.org/pub/release-115/gtf/homo_sapiens/Homo_sapiens.GRCh38.115.gtf.gz',dir='./genomes')
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
⚠️ File ./genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz already exists
🔍 Downloading data to ./genomes/Homo_sapiens.GRCh38.115.gtf.gz
✅ Download completed
'./genomes/Homo_sapiens.GRCh38.115.gtf.gz'
5) QC结果(缓存示例结构)#
以下单元格提供了**qc_result结构示例**(清洗后reads和报告的路径)。
这对于无需重新运行QC即可快速继续教程非常有用。
如果您在自己的机器上重新运行了上面的QC步骤,可以继续使用ov.alignment.fastp()生成的qc_result。
qc_result=[
{'sample': 'SRR12544419',
'clean1': 'result/fqdata/SRR12544419/SRR12544419_clean_1.fastq',
'clean2': 'result/fqdata/SRR12544419/SRR12544419_clean_2.fastq',
'json': 'result/fqdata/SRR12544419/SRR12544419.fastp.json',
'html': 'result/fqdata/SRR12544419/SRR12544419.fastp.html'},
{'sample': 'SRR12544421',
'clean1': 'result/fqdata/SRR12544421/SRR12544421_clean_1.fastq',
'clean2': 'result/fqdata/SRR12544421/SRR12544421_clean_2.fastq',
'json': 'result/fqdata/SRR12544421/SRR12544421.fastp.json',
'html': 'result/fqdata/SRR12544421/SRR12544421.fastp.html'},
{'sample': 'SRR12544529',
'clean1': 'result/fqdata/SRR12544529/SRR12544529_clean_1.fastq',
'clean2': 'result/fqdata/SRR12544529/SRR12544529_clean_2.fastq',
'json': 'result/fqdata/SRR12544529/SRR12544529.fastp.json',
'html': 'result/fqdata/SRR12544529/SRR12544529.fastp.html'},
{'sample': 'SRR12544531',
'clean1': 'result/fqdata/SRR12544531/SRR12544531_clean_1.fastq',
'clean2': 'result/fqdata/SRR12544531/SRR12544531_clean_2.fastq',
'json': 'result/fqdata/SRR12544531/SRR12544531.fastp.json',
'html': 'result/fqdata/SRR12544531/SRR12544531.fastp.html'}
]
6) 准备STAR输入列表#
STAR需要(sample, read1, read2)元组的列表。
我们从qc_result构建该列表。
from operator import itemgetter
list(map(itemgetter("sample", "clean1", "clean2"), qc_result))
[('SRR12544419',
'result/fqdata/SRR12544419/SRR12544419_clean_1.fastq',
'result/fqdata/SRR12544419/SRR12544419_clean_2.fastq'),
('SRR12544421',
'result/fqdata/SRR12544421/SRR12544421_clean_1.fastq',
'result/fqdata/SRR12544421/SRR12544421_clean_2.fastq'),
('SRR12544529',
'result/fqdata/SRR12544529/SRR12544529_clean_1.fastq',
'result/fqdata/SRR12544529/SRR12544529_clean_2.fastq'),
('SRR12544531',
'result/fqdata/SRR12544531/SRR12544531_clean_1.fastq',
'result/fqdata/SRR12544531/SRR12544531_clean_2.fastq')]
7) 使用STAR将reads比对到参考基因组#
我们使用STAR将清洗后的reads比对到GRCh38。
▲ 重要提示
首次运行将创建STAR基因组索引(基因组初始化),根据硬件和I/O性能,这可能需要约20分钟。
STAR对内存要求较高;代码注释中指出需要大量内存。
输出文件
排序后的BAM:
./result/alignment/<sample>/Aligned.sortedByCoord.out.bam(路径可能因STAR设置而异)
align_result = ov.alignment.STAR(
samples=list(map(itemgetter("sample", "clean1", "clean2"), qc_result)),
genome_dir='genomes',
genome_fasta_files="genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz",
gtf='genomes/Homo_sapiens.GRCh38.115.gtf.gz',
sjdb_overhang=100,
output_dir = './result/alignment',
jobs=8 # 1 job require at least 48GB memory
)
print(align_result)
>> /home/groups/xiaojie/steorra/env/omicverse/bin/STAR --runMode genomeGenerate --genomeDir genomes --genomeFastaFiles genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa --runThreadN 8 --sjdbGTFfile genomes/Homo_sapiens.GRCh38.115.gtf --sjdbGTFfeatureExon exon --sjdbOverhang 100
/home/groups/xiaojie/steorra/env/omicverse/bin/STAR-avx2 --runMode genomeGenerate --genomeDir genomes --genomeFastaFiles genomes/Homo_sapiens.GRCh38.dna.primary_assembly.fa --runThreadN 8 --sjdbGTFfile genomes/Homo_sapiens.GRCh38.115.gtf --sjdbGTFfeatureExon exon --sjdbOverhang 100
STAR version: 2.7.11b compiled: 2025-07-24T03:06:21+0000 :/opt/conda/conda-bld/star_1753326220084/work/source
Feb 06 17:41:00 ..... started STAR run
Feb 06 17:41:00 ... starting to generate Genome files
Feb 06 17:41:45 ..... processing annotations GTF
8) 比对结果(缓存示例结构)#
下一个单元格提供了示例align_result列表(样本 → BAM路径)。
即使在本教程环境中跳过重新运行STAR,您也可以继续进行定量步骤。
align_result=[
{'sample':'SRR12544419',
'bam':'result/alignment/SRR12544419/Aligned.sortedByCoord.out.bam'},
{'sample':'SRR12544421',
'bam':'result/alignment/SRR12544421/Aligned.sortedByCoord.out.bam'},
{'sample':'SRR12544529',
'bam':'result/alignment/SRR12544529/Aligned.sortedByCoord.out.bam'},
{'sample':'SRR12544531',
'bam':'result/alignment/SRR12544531/Aligned.sortedByCoord.out.bam'},
]
9) 使用featureCounts进行基因水平定量#
我们使用featureCounts(通过OmicVerse封装)将reads定量到基因,生成原始计数矩阵。
输出
count_result:一个基因 × 样本(原始计数)的pandas DataFrame,当gene_mapping=True时可选地映射到基因符号。
count_result = ov.alignment.featureCount(
bam_items=list(map(itemgetter("sample", "bam"), align_result)),
gtf='genomes/Homo_sapiens.GRCh38.115.gtf.gz',
output_dir = './result/count',
gene_mapping=True,
gene_name_field="gene_name",
jobs=8
)
print(count_result)
SRR12544419 SRR12544421 SRR12544529 SRR12544531
gene_id
PRDM16 2 0 0 0
ENSG00000284616 0 0 0 0
ENSG00000260972 0 0 0 0
EEF1DP6 0 0 0 0
LINC01646 0 0 0 0
... ... ... ... ...
ENSG00000307722 0 0 0 0
ENSG00000310401 0 0 0 0
ENSG00000302039 0 2 24 0
ENSG00000309831 2 6 0 1
ENSG00000309258 0 0 0 0
[78899 rows x 4 columns]
count_result.head()
| SRR12544419 | SRR12544421 | SRR12544529 | SRR12544531 | |
|---|---|---|---|---|
| gene_id | ||||
| PRDM16 | 2 | 0 | 0 | 0 |
| ENSG00000284616 | 0 | 0 | 0 | 0 |
| ENSG00000260972 | 0 | 0 | 0 | 0 |
| EEF1DP6 | 0 | 0 | 0 | 0 |
| LINC01646 | 0 | 0 | 0 | 0 |
10) 创建DE对象(pyDEG)#
我们从计数矩阵构建OmicVerse的pyDEG对象,用于下游DE分析和绘图。
data = count_result
dds=ov.bulk.pyDEG(data)
11) 基础清理#
我们删除重复的基因条目(如有)。
下游步骤假设索引唯一标识基因。
dds.drop_duplicates_index()
print('... drop_duplicates_index success')
... drop_duplicates_index success
12) 差异表达(DE)#
定义处理组/对照组样本列表并运行DE分析。OmicVerse支持多种DE方法,包括:
ttestedgepylimmaDEseq2
▲ 重要提示(DESeq2)
如果使用DEseq2,请确保输入矩阵为原始整数计数,并避免事先应用外部归一化(DESeq2在内部估算大小因子)。
disease_groups = ['SRR12544419','SRR12544421']
healthy_groups = ['SRR12544529','SRR12544531']
result=dds.deg_analysis(disease_groups,healthy_groups,method='DEseq2')
⚙️ You are using DEseq2 method for differential expression analysis.
⏰ Start to create DeseqDataSet...
logres_prior=1.028669461109852, sigma_prior=0.25
Log2 fold change & Wald test p-value: condition Treatment vs Control
baseMean log2FoldChange lfcSE stat \
gene_id
S100A9 551268.969994 -1.996395 0.567251 -3.519423
S100A8 470875.870644 -1.860539 0.580843 -3.203169
MT-RNR2 379360.147119 -1.073334 0.462907 -2.318684
HBB 255919.879002 -3.412494 0.550731 -6.196301
B2M 224562.599264 -0.016376 0.477808 -0.034274
... ... ... ... ...
RN7SKP39 0.000000 NaN NaN NaN
RNU4ATAC6P 0.000000 NaN NaN NaN
TPRKBP1 0.000000 NaN NaN NaN
RNU7-107P 0.000000 NaN NaN NaN
ENSG00000271220 0.000000 NaN NaN NaN
pvalue padj
gene_id
S100A9 4.324866e-04 0.036766
S100A8 1.359241e-03 0.074803
MT-RNR2 2.041219e-02 0.334090
HBB 5.780550e-10 0.000002
B2M 9.726589e-01 0.995301
... ... ...
RN7SKP39 NaN NaN
RNU4ATAC6P NaN NaN
TPRKBP1 NaN NaN
RNU7-107P NaN NaN
ENSG00000271220 NaN NaN
[78899 rows x 6 columns]
✅ Differential expression analysis completed.
13) 过滤低表达基因#
简单的过滤可以减少噪音并提高可解释性。
dds.result=dds.result.loc[dds.result['log2(BaseMean)']>1]
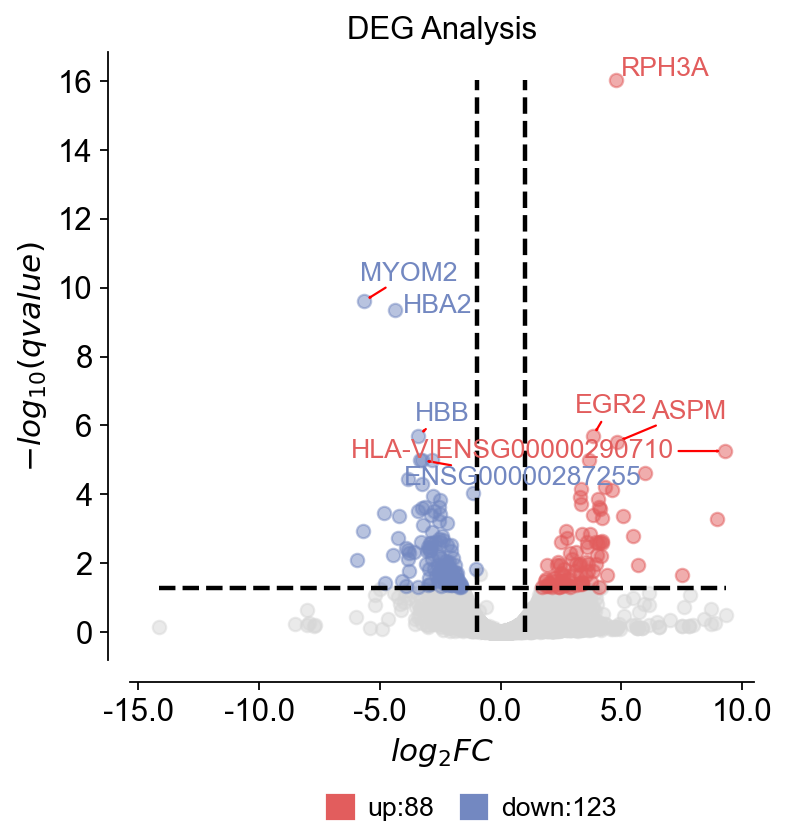
14) 设置阈值并检查顶部DE基因#
我们设置倍数变化/p值阈值,并提取显著DEG的前10名进行可视化。
import matplotlib.pyplot as plt
# fc_treshold = -1 means automatically calculate
dds.foldchange_set(fc_threshold=1,pval_threshold=0.05,logp_max=20)
res = dds.result.copy()
sig = res[(res["qvalue"] < 0.05) & (res["log2FC"].abs() > 0.5)&(res["log2(BaseMean)"]<20)].sort_values(["qvalue","log2FC"], ascending=[True, False])
gene_list = sig[["log2FC","qvalue"]].head(10).index.to_list() # Top10 significant DEGs
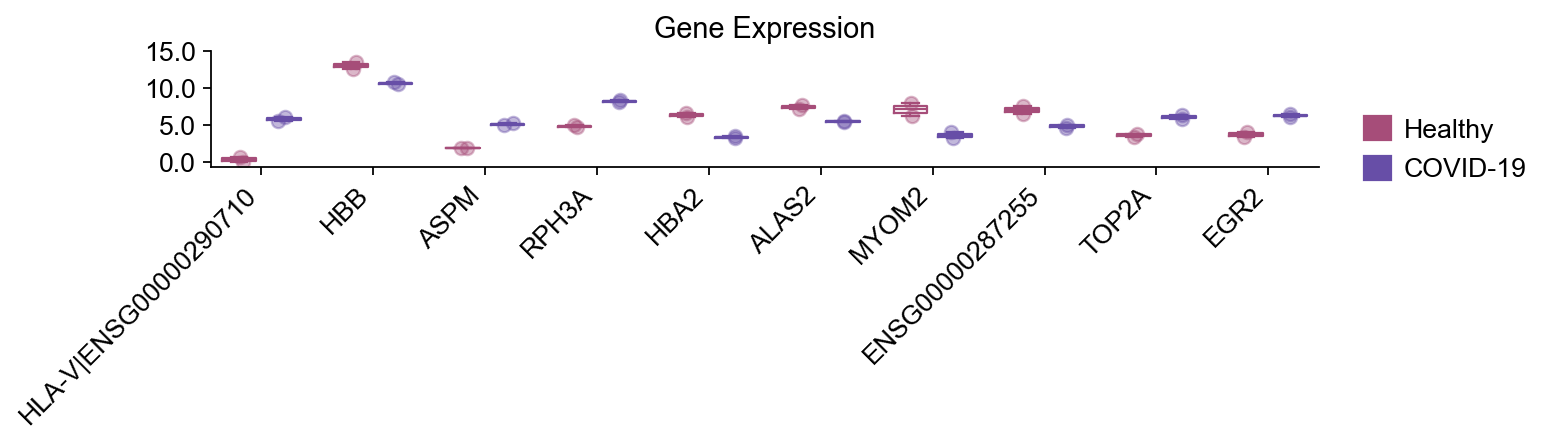
print("Top10 significant DEGs:",gene_list)
... Fold change threshold: 1
Top10 significant DEGs: ['RPH3A', 'MYOM2', 'HBA2', 'EGR2', 'HBB', 'ASPM', 'HLA-V|ENSG00000290710', 'TOP2A', 'ALAS2', 'ENSG00000287255']
15) 可视化DE结果#
我们生成:
汇总DE结果的火山图
所选基因的箱线图(顶部候选基因)
提示:dds.plot_boxplot()有助于对各组中单个基因的行为进行合理性检查。
dds.plot_volcano(title='DEG Analysis',figsize=(5,5),
plot_genes_num=8,plot_genes_fontsize=12,)
dds.plot_boxplot(genes= gene_list,
treatment_groups=disease_groups,
treatment_name='COVID-19',control_name='Healthy',
control_groups=healthy_groups,figsize=(10,3),fontsize=12,
legend_bbox=(1.2,0.55))
plt.xticks(rotation=45, ha="right")
plt.tight_layout()
🌋 Volcano Plot Analysis:
Total genes: 25211
↗️ Upregulated genes: 88
↘️ Downregulated genes: 123
➡️ Non-significant genes: 25000
🎯 Total significant genes: 211
log2FC range: -14.16 to 9.33
qvalue range: 9.31e-17 to 1.00e+00
⚙️ Current Function Parameters:
Data columns: pval_name='qvalue', fc_name='log2FC'
Thresholds: pval_threshold=0.05, fc_max=1, fc_min=-1
Plot size: figsize=(5, 5)
Gene labels: plot_genes_num=8, plot_genes_fontsize=12
Custom genes: None (auto-select top genes)
💡 Parameter Optimization Suggestions:
▶ Wide fold change range detected:
Current: fc_max=1, fc_min=-1
Suggested: fc_max=2.6, fc_min=-2.8
📋 Copy-paste ready function call:
ov.pl.volcano(result, fc_max=2.6, fc_min=-2.8)
────────────────────────────────────────────────────────────
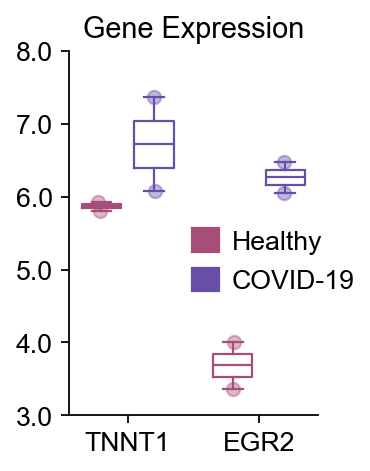
16) 额外的箱线图示例#
一个简洁的示例,展示如何手动指定来自其他上游分析步骤的基因列表进行绘图。
dds.plot_boxplot(genes=['TNNT1','EGR2'],
treatment_groups=disease_groups,
treatment_name='COVID-19',control_name='Healthy',
control_groups=healthy_groups,figsize=(2,3),fontsize=12,
legend_bbox=(1.2,0.55))
(<Figure size 160x240 with 1 Axes>,
<Axes: title={'center': 'Gene Expression'}>)
后续步骤(可选)#
从此处开始,典型的下游bulk RNA-seq分析包括:
对上调/下调DEG进行功能富集(如GO / KEGG / Reactome)
蛋白质-蛋白质相互作用(PPI)网络构建和可视化
通路水平评分和报告
一旦确定了研究中的DEG,您可以通过添加这些部分来扩展本Notebook。