Metabolite cell-cell communication in a tumour microenvironment#
The companion tutorial scored metabolism within each cell. Here we
ask the question that needs the whole ecosystem: in a head & neck
tumour, which cell types feed which — which metabolites flow from
which sender to which receiver? ov.single.MetaboliteCCC wraps
MEBOCOST to infer metabolite-mediated cell-cell communication, and
reuses the ov.pl.ccc_* communication plots.
Part.1 The idea behind MEBOCOST#
A metabolite-mediated communication event has two ends:
a sender cell type that makes a metabolite available — read out as high expression of its synthesising / exporting enzymes;
a receiver cell type that takes it up or senses it — high expression of a matching sensor: a transporter, a cell-surface receptor or a nuclear receptor.
MEBOCOST estimates per-cell-type metabolite abundance from enzyme expression, pairs metabolites with sensors through a curated database, and scores every sender to receiver event against a permutation null. Tumour metabolism is not cell-autonomous — malignant cells, fibroblasts and immune cells feed and starve one another, and this is how that traffic is mapped.
import omicverse as ov
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
🚫 No GPU devices found (CUDA/MPS/ROCm/XPU)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
Part.2 The HNSC tumour atlas#
The same Puram et al. 2017 head & neck cancer atlas (GSE103322) used by the per-cell metabolism tutorial — 5,578 cells, malignant plus the stromal and immune microenvironment.
adata = ov.datasets.metabolism_hnsc()
adata.obs['celltype'].value_counts()
🔍 Downloading data to ./data/hnsc_puram2017_full.h5ad
⚠️ File ./data/hnsc_puram2017_full.h5ad already exists
celltype
Malignant 2215
Fibroblast 1440
T cell 1237
Endothelial 260
B cell 138
Mast 120
Macrophage 98
Dendritic 51
myocyte 19
Name: count, dtype: int64
Part.3 Infer metabolite communication#
ov.single.MetaboliteCCC takes the cell-type column as the unit of
communication. run() estimates metabolite abundance, pairs
metabolites with sensors and runs the permutation test.
min_cell_number drops cell types too small to estimate from, and
n_shuffle sizes the null (1000 for a publication; 100 here to keep
the tutorial quick).
mccc = ov.single.MetaboliteCCC(adata, group_key='celltype')
mccc.run(n_shuffle=100, min_cell_number=30, verbose=False)
<omicverse.single._metabolism.MetaboliteCCC at 0x7f594e709780>
# the strongest metabolite -> sensor communication events
mccc.result.nlargest(10, 'Commu_Score')[
['Sender', 'Receiver', 'Metabolite_Name', 'Sensor',
'Commu_Score', 'permutation_test_fdr']]
| Sender | Receiver | Metabolite_Name | Sensor | Commu_Score | permutation_test_fdr | |
|---|---|---|---|---|---|---|
| 73 | Mast | Malignant | L-Glutamine | SLC3A2 | 20.723214 | 0.0 |
| 37 | Macrophage | Malignant | L-Glutamine | SLC3A2 | 19.115429 | 0.0 |
| 76 | Mast | Macrophage | L-Glutamine | SLC3A2 | 14.312806 | 0.0 |
| 74 | Mast | B cell | L-Glutamine | SLC3A2 | 14.117096 | 0.0 |
| 72 | Mast | Fibroblast | L-Glutamine | SLC38A2 | 13.493294 | 0.0 |
| 40 | Macrophage | Macrophage | L-Glutamine | SLC3A2 | 13.202364 | 0.0 |
| 74 | Mast | B cell | L-Glutamine | SLC38A2 | 13.141793 | 0.0 |
| 38 | Macrophage | B cell | L-Glutamine | SLC3A2 | 13.021839 | 0.0 |
| 10 | Malignant | Malignant | L-Glutamine | SLC3A2 | 12.874932 | 0.0 |
| 52 | Endothelial | Dendritic | Farnesyl pyrophosphate | GPR183 | 12.629885 | 0.0 |
Part.4 The communication network#
to_comm_adata() converts the MEBOCOST result into a communication
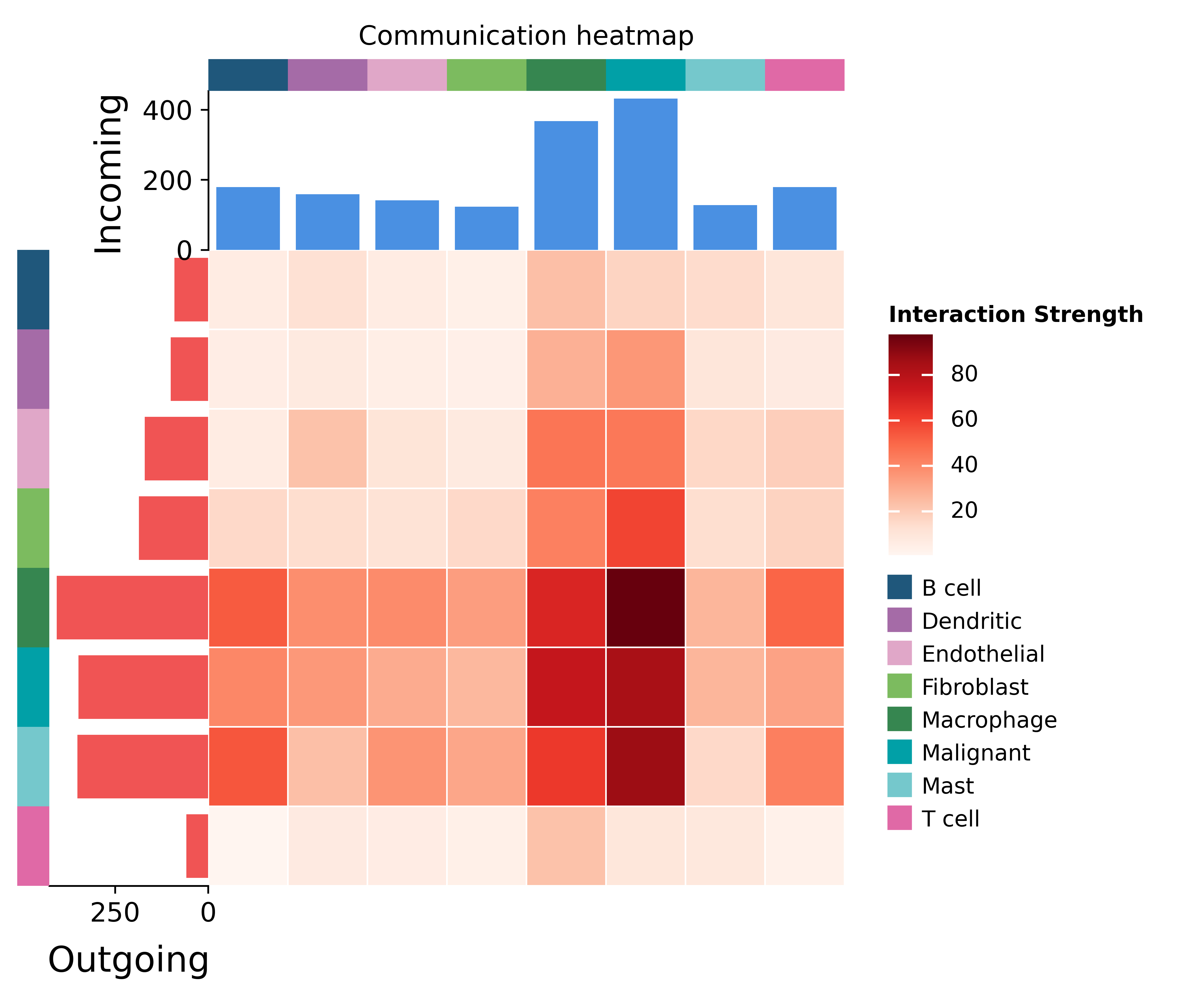
AnnData — the schema the ov.pl.ccc_* plots consume. The heatmap
sums communication strength over every sender to receiver pair; a
bright row is a metabolite source, a bright column a sink.
comm = mccc.to_comm_adata()
fig, ax = ov.pl.ccc_heatmap(comm, plot_type='heatmap')
fig
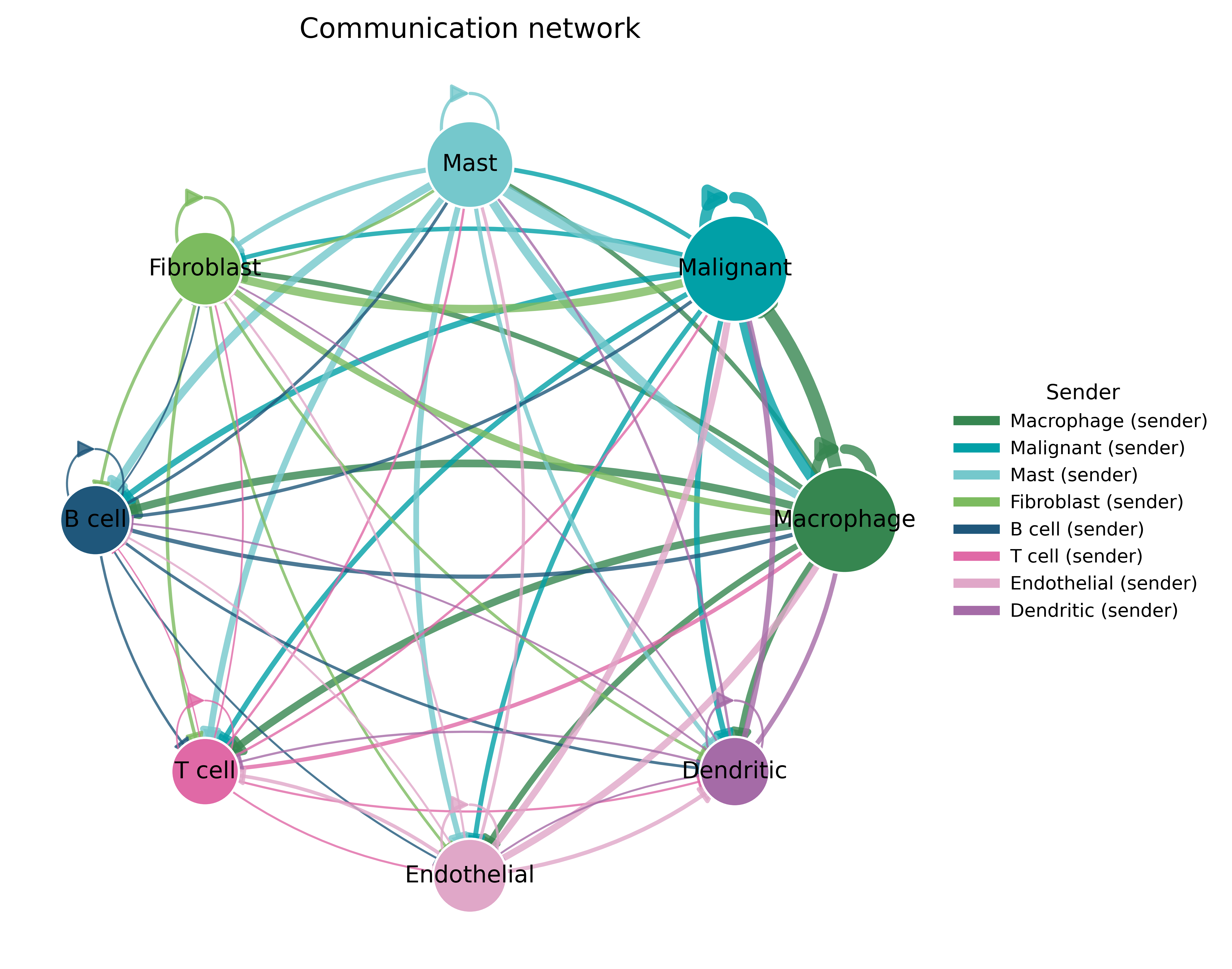
The same traffic as a directed network — arrows run from metabolite senders to receivers, weighted by communication strength.
fig, ax = ov.pl.ccc_network_plot(comm, plot_type='circle')
fig
Part.5 Drill into specific metabolite-sensor channels#
The aggregated views in Part.4 confirm that signal is flowing — they
do not say what is in the bottle. Every row of mccc.result names a
specific (metabolite, sensor) pair, and to_comm_adata() keeps that
resolution: metabolites land in the gene_a / ligand slot of the
comm AnnData, sensors in gene_b / receptor. That makes the entire
interaction-level ov.pl.ccc_* family — the same plots used by the
LIANA ligand-receptor tutorial — available on a metabolite-CCC result.
The default interaction-level view is the LIANA-style dot plot. Rows
are metabolite → sensor pairs, columns are receivers, facets are
senders. Dot size encodes −log10(FDR), color encodes the score.
fig, ax = ov.pl.ccc_heatmap(
comm, plot_type='dot', display_by='interaction',
top_n=10, pvalue_threshold=0.05,
figsize=(16, 6.5), show=False)
fig
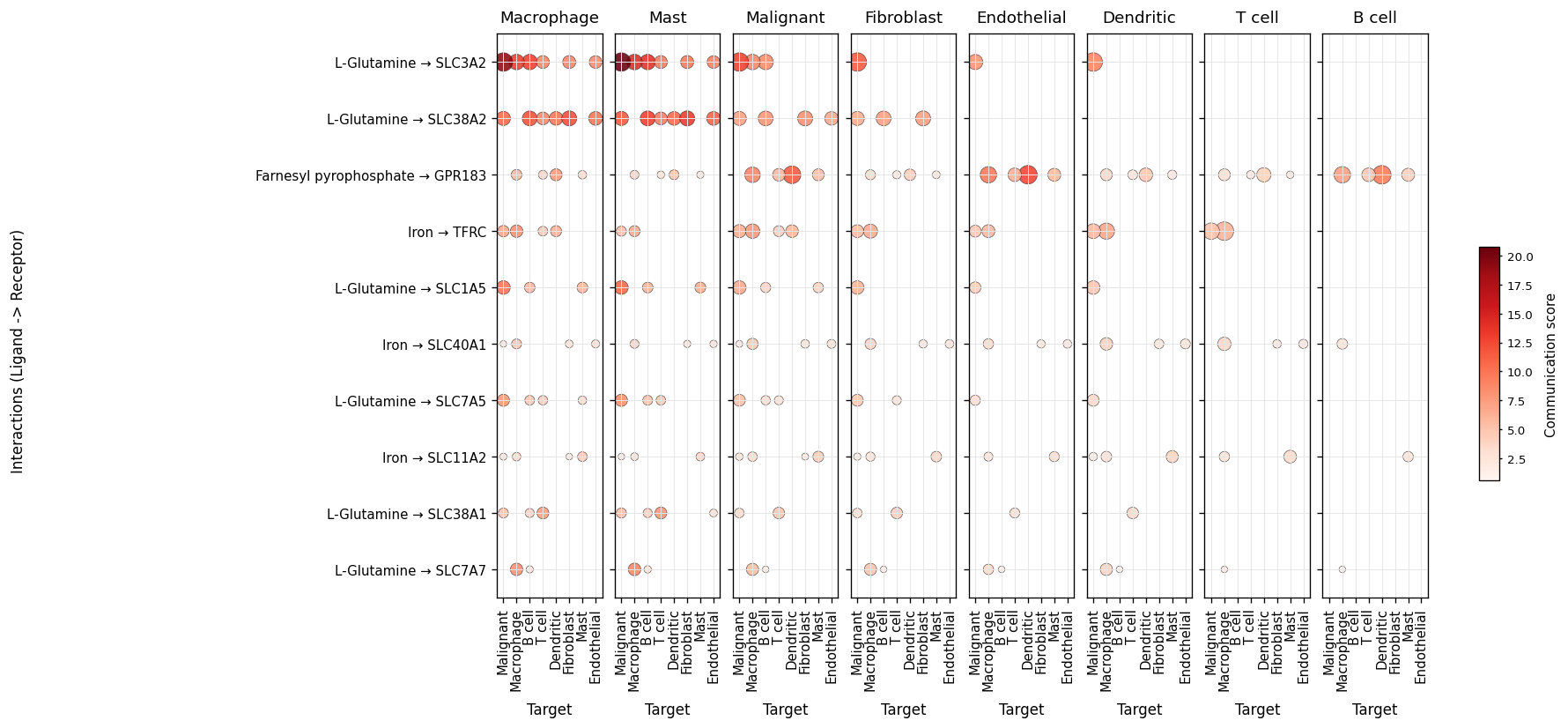
Sender / receiver focus#
Once a cell type of interest is in mind, restrict the same plot to
that role. sender_use=['Mast'] asks “of everything Mast cells put
into the system, what dominates?”. receiver_use=['Malignant'] asks
the symmetric question — who and what is feeding the tumour.
fig, ax = ov.pl.ccc_heatmap(
comm, plot_type='dot', display_by='interaction',
sender_use=['Mast'], top_n=10, pvalue_threshold=0.05,
figsize=(5.5, 5), show=False)
fig
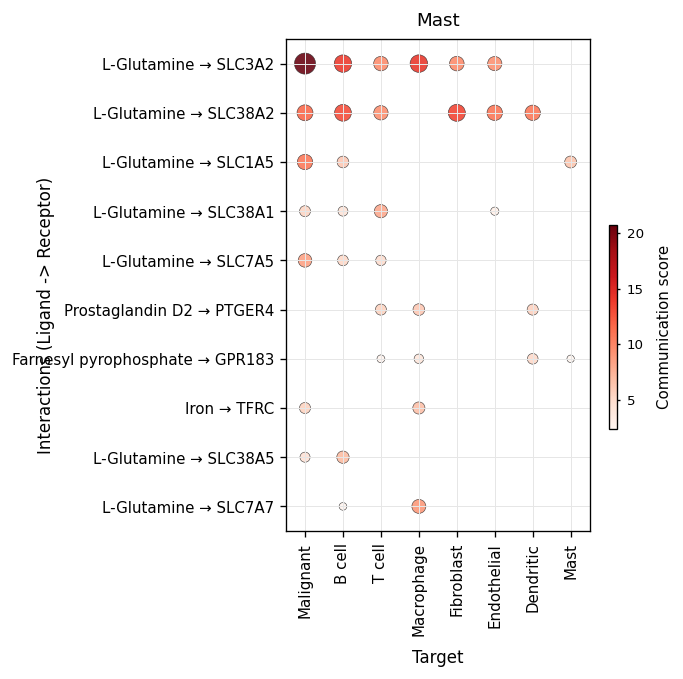
fig, ax = ov.pl.ccc_heatmap(
comm, plot_type='dot', display_by='interaction',
receiver_use=['Malignant'], top_n=10, pvalue_threshold=0.05,
figsize=(10, 5), show=False)
fig
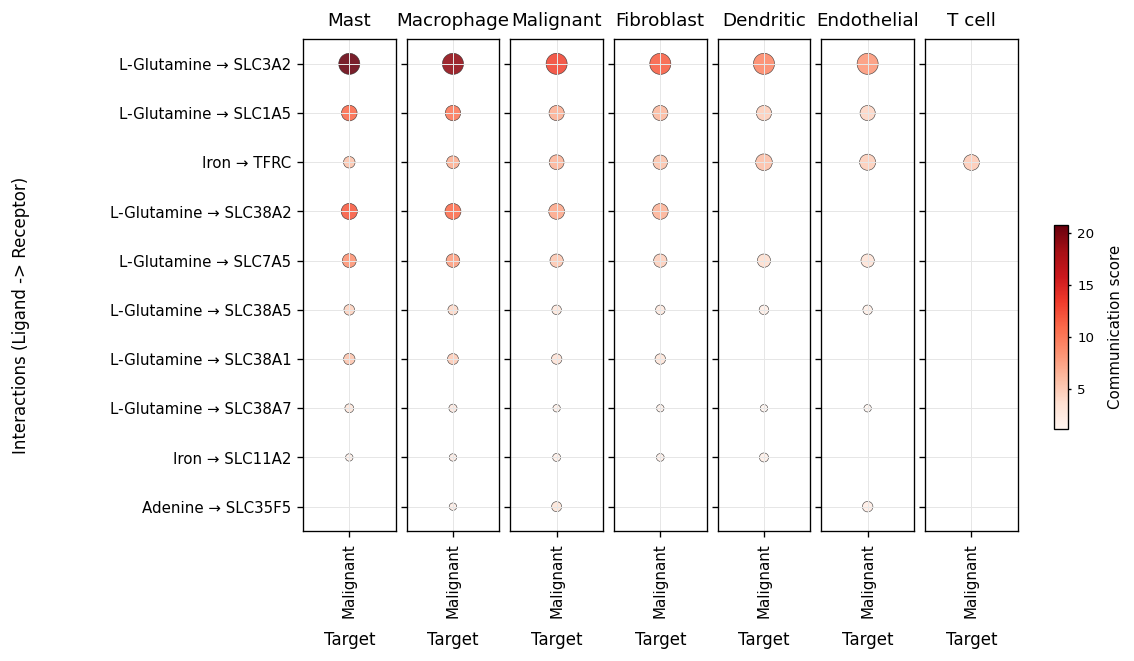
Part.6 Production vs sensing — tile + bar#
tile puts the two ends of the channel side by side: left is what
the sender produces (metabolite abundance), right is what the
receiver expresses (sensor expression). A channel is a real signal
only when both panels light up for the same (metabolite → sensor)
row.
bar then collapses the matrix to a flat ranking — the strongest
individual interactions across the system, useful for picking the
next channel to drill into.
fig, ax = ov.pl.ccc_heatmap(
comm, plot_type='tile', display_by='interaction',
top_n=10, pvalue_threshold=0.05,
figsize=(7, 4), show=False)
fig
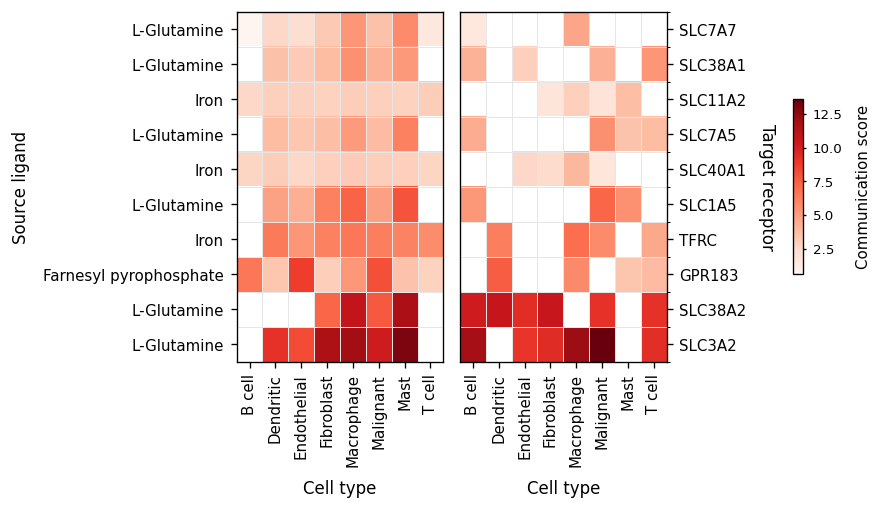
fig, ax = ov.pl.ccc_stat_plot(
comm, plot_type='bar', display_by='interaction',
group_by='interaction', top_n=12,
figsize=(6, 5), show=False)
fig
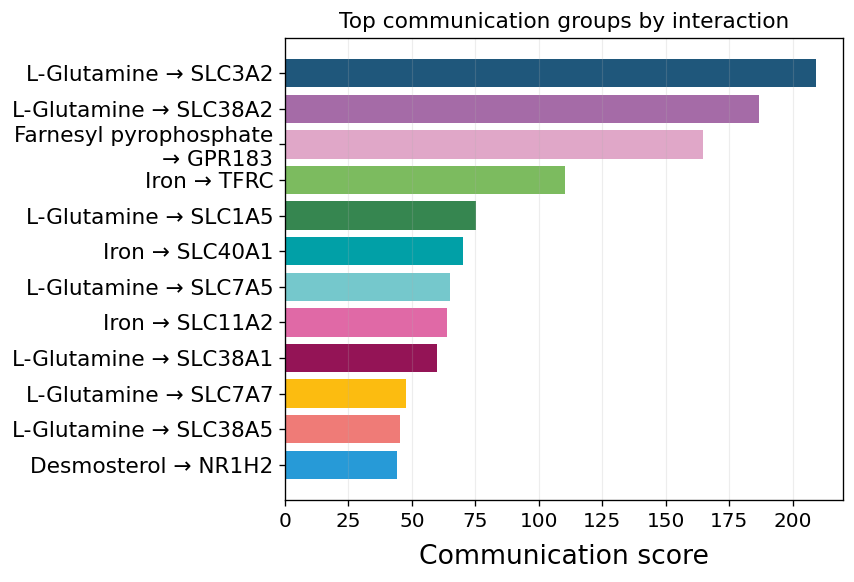
Part.7 Metabolite classes shape the landscape#
HMDB’s chemical classification is joined onto the comm AnnData as
var['classification'] (sub_class — e.g. “Amino acids, peptides, and
analogues”, “Carbohydrates and carbohydrate conjugates”) and
var['classification_super'] (super_class). Aggregating by class
turns the long list of individual channels into a small set of
metabolic programs: which broad class of metabolites is most
active across the system.
pathway_summary ranks classes by total communication strength and
annotates each bar with n_significant / n_active cell-pairs — a
single-glance way to see which class of metabolites carries the most
traffic and how widely it reaches.
fig, ax = ov.pl.ccc_stat_plot(
comm, plot_type='pathway_summary',
top_n=10, figsize=(7, 5), show=False)
fig
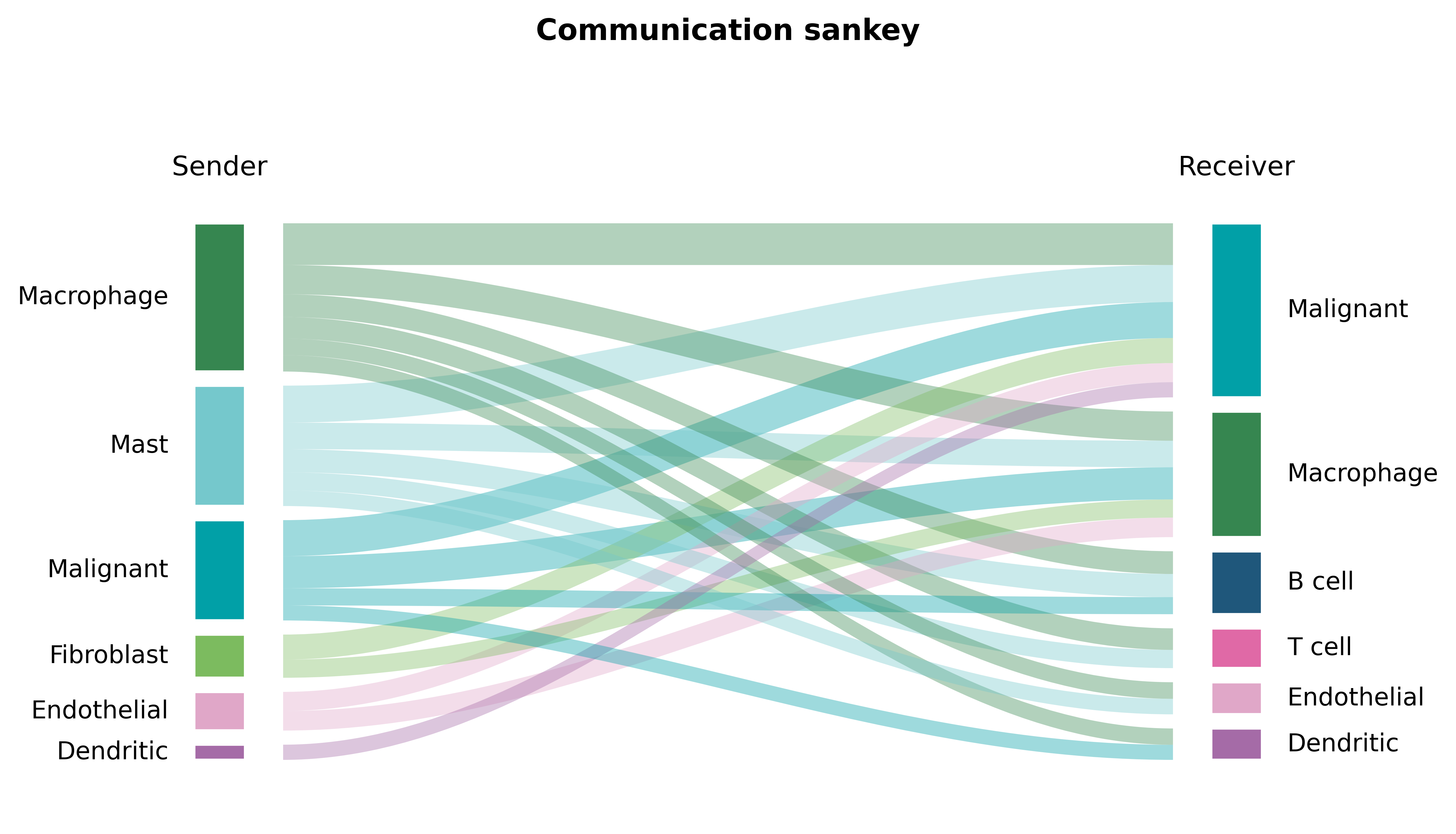
Part.8 The dominant metabolite channels#
A Sankey view follows the flow end-to-end: which cell types pour the most metabolite signal into which receivers.
fig, ax = ov.pl.ccc_stat_plot(comm, plot_type='sankey')
fig
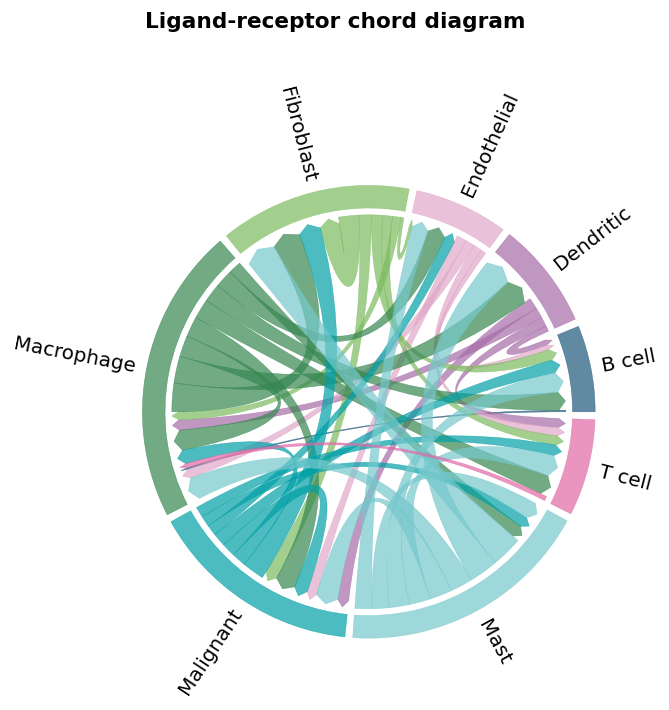
Part.9 A closer look — L-Glutamine#
Reading the top events again: they nearly all converge on one channel — malignant cells receiving L-glutamine. The sensors are SLC3A2 (CD98hc), SLC1A5 and SLC38A2 — amino-acid transporters, not receptors — so MEBOCOST is reading metabolite uptake. This is textbook cancer biology: tumour cells are “glutamine-addicted”, and the analysis names who pays for the habit.
Filter the comm AnnData to the L-Glutamine channels and draw a sender-receiver chord — every L-Glutamine → sensor pair in one view.
gln_interactions = [v for v in comm.var_names if v.startswith('L-Glutamine')]
fig, ax = ov.pl.ccc_network_plot(
comm, plot_type='lr_chord',
pair_lr_use=gln_interactions,
figsize=(6, 6), show=False)
fig
recv = mccc.result[mccc.result['Receiver'] == 'Malignant']
(recv.groupby('Metabolite_Name')['Commu_Score'].sum()
.sort_values(ascending=False).head(10))
Metabolite_Name
L-Glutamine 246.935687
Iron 53.167821
Leukotriene B4 12.561020
gamma-Aminobutyric acid 9.449661
L-Serine 8.482404
3-Hydroxybutyric acid 7.552795
Adenine 7.424385
D-Mannose 7.241953
Desmosterol 6.615091
Cholesterol 6.119173
Name: Commu_Score, dtype: float64
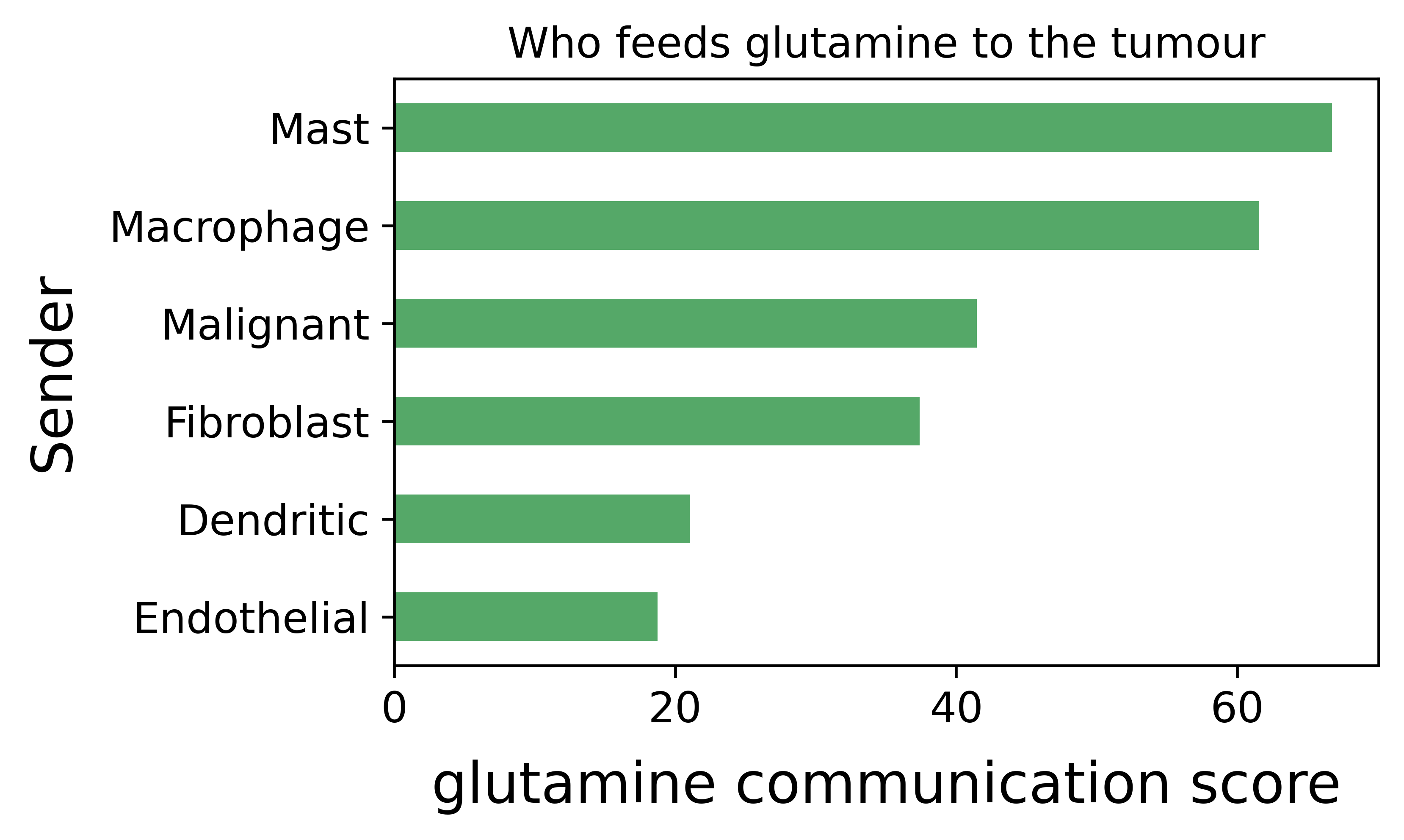
import matplotlib.pyplot as plt
plt.close('all') # clear figures left open by the ccc_* plots
gln = mccc.result.query("Receiver == 'Malignant' "
"and Metabolite_Name == 'L-Glutamine'")
supply = gln.groupby('Sender')['Commu_Score'].sum().sort_values()
supply.plot.barh(color='#55A868', figsize=(5, 3),
xlabel='glutamine communication score',
title='Who feeds glutamine to the tumour')
plt.show()
Recap#
ov.single.MetaboliteCCC lifts the metabolism analysis from the
single cell to the metabolic ecology of the tumour. On this head &
neck cancer atlas it recovers a real dependency: the malignant
compartment is a metabolite sink, and mast cells and macrophages
are the chief suppliers of the L-glutamine that glutamine-addicted
tumour cells consume.
The comm AnnData built by MetaboliteCCC.to_comm_adata() keeps every
(metabolite, sensor) pair as its own column — metabolites in the
ligand slot, sensors in the receptor slot, HMDB sub/super-class
in the classification slot. That schema is what unlocks the full
interaction-level ov.pl.ccc_* family in this tutorial: the dot /
tile / bar / pathway-bubble / chord views are the same plots the
LIANA tutorial uses on protein ligand-receptor pairs, applied here
to metabolite channels.
Paired with the per-cell scMetabolism / scFEA analysis, it completes the picture — metabolic state within cells and metabolite exchange between them.