Missing values in proteomics — diagnosis & imputation#
Missing values are the defining problem of quantitative proteomics. A typical label-free DDA experiment leaves 30-50 % of the intensity matrix empty — far more than RNA-seq, and crucially the gaps are not random. How you fill those gaps changes every downstream number: fold-changes, p-values, PCA structure, and the final biological conclusions. This tutorial is a deep dive into getting it right.
The two missingness mechanisms#
A value can be missing for two fundamentally different reasons:
Mechanism |
Cause |
Where the true value sits |
|---|---|---|
MCAR / MAR — missing (completely) at random |
instrument hiccup, a mis-aligned MS1 feature, a stochastic identification failure |
anywhere in the protein’s normal range |
MNAR — missing not at random |
the protein was genuinely below the detection limit of the instrument |
low — at or under the limit of detection (left-censored) |
This distinction is the whole game. An MNAR value is informative: its absence tells you the protein is low-abundance. If you impute it with the protein’s mean (which is what KNN / mean-imputation effectively do), you invent signal that was never there — a low-abundance protein suddenly looks mid-abundance, and a real “present vs absent” difference between conditions is erased. Conversely, if a value is truly MCAR and you impute it with a low detection-limit draw, you fabricate a downward bias.
So there is no universally best imputer. The right method depends on the mechanism, and the mechanism must be diagnosed from the data.
What this tutorial does#
Load a real ProteomeXchange dataset and quantify its missingness.
Diagnose the mechanism — is this MNAR?
Tour the imputer zoo and see how each one places its imputed values.
Benchmark imputers objectively by artificially masking known values and measuring reconstruction error on this real data.
Show that the imputer choice materially changes the biology (the DE result).
Arrive at a principled, mechanism-aware recommended strategy.
The benchmarking (section 4) is the substance: it turns “which imputer?” from an opinion into a measurement.
Imports#
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import omicverse as ov
import pyimputelcmd as pl
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
🚫 No GPU devices found (CUDA/MPS/ROCm/XPU)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
1. The dataset & its missingness#
We use PXD000438, a real label-free DDA dataset from ProteomeXchange. It
contains 3709 proteins quantified across 12 samples, organised into 4 groups
(092, 441, 561, 691) with 3 replicates each. adata.X holds the raw
MS1 intensities with genuine, left-censored missing values encoded as NaN —
exactly the kind of data this tutorial is about.
In omicverse’s proteomics convention the AnnData is samples (obs) ×
proteins (var).
adata = ov.datasets.protein_pxd000438()
adata
🔍 Downloading data to ./data/protein_pxd000438.h5ad
✅ Download completed
AnnData object with n_obs × n_vars = 12 × 3709
obs: 'group'
uns: 'source'
# group layout: 4 conditions, 3 replicates each
adata.obs['group'].value_counts().sort_index()
group
092 3
441 3
561 3
691 3
Name: count, dtype: int64
missing_pattern summarises the gaps three ways: per-protein, per-sample,
and overall. The overall figure tells us how big the problem is; the per-sample
spread tells us whether one run was unusually bad.
mp = ov.protein.missing_pattern(adata)
print(f"Overall missing fraction : {mp['overall']:.1%}")
print(f"Proteins fully observed : {(mp['protein_missing_frac'] == 0).sum()} / {adata.n_vars}")
print(f"Proteins >50% missing : {(mp['protein_missing_frac'] > 0.5).sum()} / {adata.n_vars}")
Overall missing fraction : 40.8%
Proteins fully observed : 1142 / 3709
Proteins >50% missing : 1515 / 3709
# per-sample missingness: is any single run an outlier?
mp['sample_missing_frac'].round(3)
092.1 0.189
092.2 0.360
092.3 0.364
441.1 0.442
441.2 0.452
441.3 0.449
561.1 0.456
561.2 0.434
561.3 0.434
691.1 0.449
691.2 0.439
691.3 0.435
Name: missing_frac, dtype: float64
Roughly 41 % of the matrix is empty — and it is spread fairly evenly
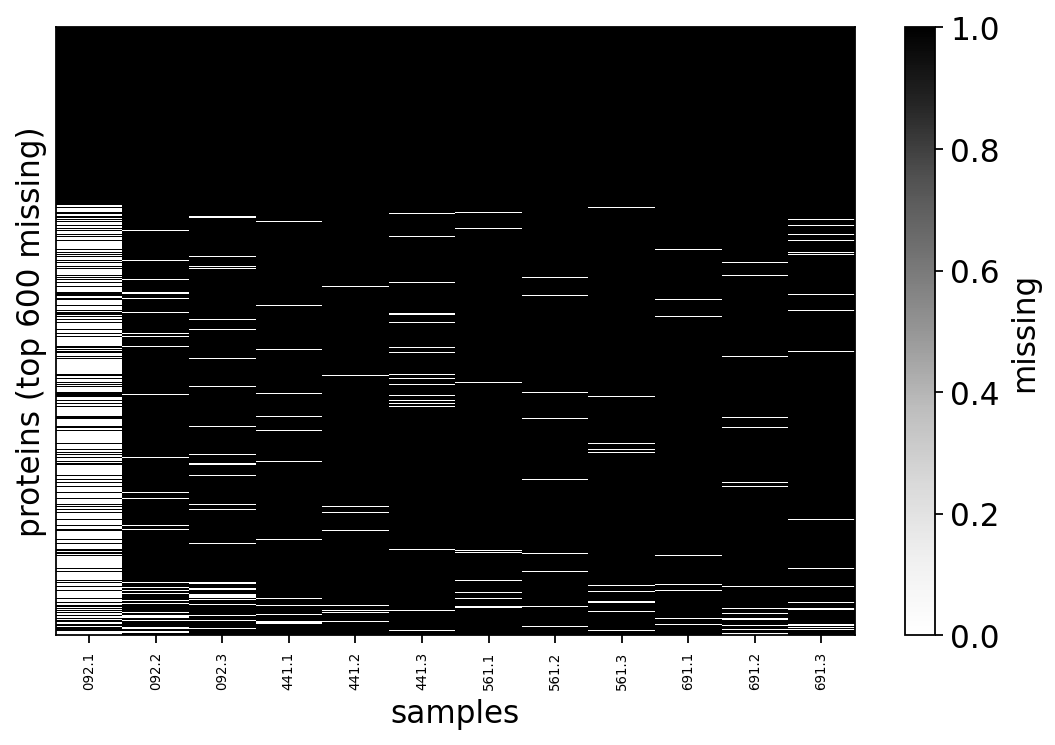
across samples (no single failed run to discard). The missing_pattern_plot
below renders the matrix itself: black = missing. The decisive visual is the
structure — gaps cluster into whole rows (proteins missing everywhere) rather
than scattering uniformly, the first hint that missingness tracks abundance.
ov.protein.missing_pattern_plot(adata, max_proteins=600)
plt.show()
2. Is it MNAR?#
The single most informative diagnostic for proteomics missingness is a scatter of, for each protein, its mean observed abundance against its fraction of missing values.
If missingness were MCAR, the cloud would be flat — low- and high-abundance proteins would be missing equally often.
If missingness is MNAR / left-censored, the cloud slopes downward: low-abundance proteins are missing far more often, because they sit near the detection limit.
We compute means/fractions across the protein axis. Because abundances span orders of magnitude, we work in log2 space for the abundance axis.
X = adata.X # samples x proteins
miss_frac = np.isnan(X).mean(axis=0) # per-protein missing fraction
mean_abund = np.log2(np.nanmean(X, axis=0)) # per-protein mean log2 abundance
keep = np.isfinite(mean_abund) # drop all-missing proteins
r = np.corrcoef(mean_abund[keep], miss_frac[keep])[0, 1]
print(f"corr(mean abundance, missing fraction) = {r:.3f}")
corr(mean abundance, missing fraction) = -0.609
fig, ax = plt.subplots(figsize=(5.4, 4.2))
ax.scatter(mean_abund[keep], miss_frac[keep], s=6, alpha=0.25, c='#1f77b4')
# binned trend line
bins = np.linspace(np.nanmin(mean_abund[keep]), np.nanmax(mean_abund[keep]), 20)
idx = np.digitize(mean_abund[keep], bins)
trend = [np.nanmean(miss_frac[keep][idx == b]) for b in range(1, len(bins))]
ax.plot(0.5 * (bins[:-1] + bins[1:]), trend, '-o', c='#d62728', ms=4, label='binned mean')
ax.set_xlabel('mean protein abundance (log2 intensity)')
ax.set_ylabel('fraction of values missing')
ax.set_title(f'Abundance vs missingness (r = {r:.2f})')
ax.legend(frameon=False)
plt.show()
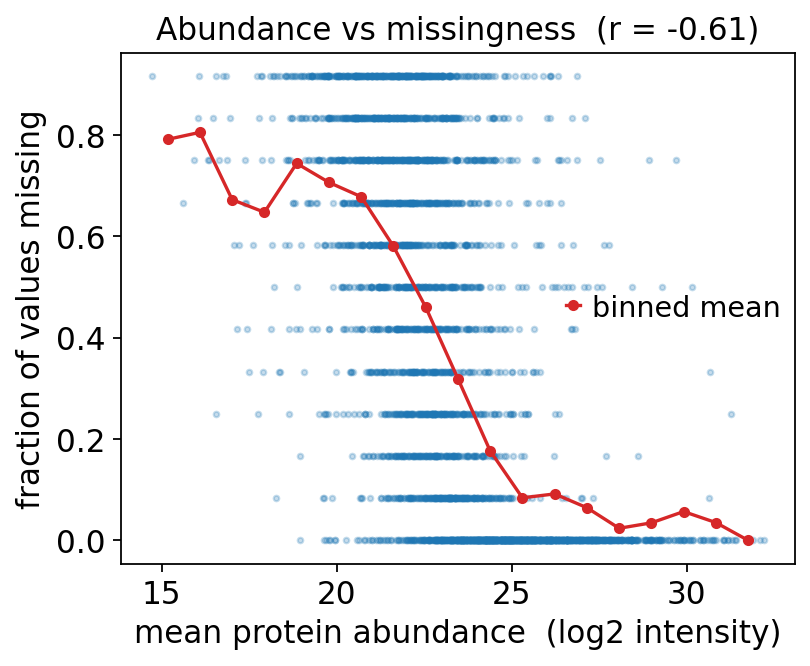
The strong negative correlation (around -0.6) and the clear downward trend line are unambiguous: low-abundance proteins are missing far more often than high-abundance ones. This is the signature of left-censored, MNAR missingness. The detection limit of the mass spectrometer is the cause.
model_selector formalises this per protein. It fits the global intensity
distribution, estimates a censoring threshold, and writes a boolean
adata.var['is_mcar'] flag (True = looks MCAR, False = looks MNAR /
left-censored). It returns the per-protein mask and the threshold.
ad = adata.copy()
ov.protein.normalize(ad, method='median', log2=True) # selector works in log2 space
mcar_mask, threshold = ov.protein.model_selector(ad)
n_mcar = int(ad.var['is_mcar'].sum())
print(f"Censoring threshold (log2 intensity) : {threshold:.2f}")
print(f"Proteins flagged MCAR-like : {n_mcar} / {ad.n_vars}")
print(f"Proteins flagged MNAR-like : {ad.n_vars - n_mcar} / {ad.n_vars}")
Censoring threshold (log2 intensity) : 19.22
Proteins flagged MCAR-like : 3386 / 3709
Proteins flagged MNAR-like : 323 / 3709
Two facts now coexist and both are true:
The global abundance-vs-missingness correlation proves the dataset as a whole is dominated by MNAR / left-censoring.
The per-protein
model_selectortest, with only 12 samples per protein, has very little power to localise the mechanism to individual proteins, so it conservatively labels most proteins MCAR-like.
The practical reading: treat this dataset as predominantly MNAR — the global evidence is decisive — while using the per-protein flag only as a soft hint. The benchmark in section 4 will confirm this empirically rather than by assumption.
3. The imputer zoo#
omicverse (via pyimputelcmd) ships three families of imputers. Knowing which
family an imputer belongs to is knowing what assumption it bakes in:
Method |
Family |
Assumption / behaviour |
|---|---|---|
|
MNAR / left-censored |
deterministic low quantile of each sample — fills near the detection limit |
|
MNAR / left-censored |
random draws from a Gaussian shifted down toward the detection limit |
|
MNAR / left-censored |
quantile-regression imputation of left-censored data — truncated-normal draws below the censoring point |
|
MAR |
average of the k most similar proteins — fills inside the observed range |
|
MAR |
maximum-likelihood (EM) estimate under a multivariate normal |
|
MAR |
low-rank reconstruction from the dominant components |
|
naive |
fills with 0 — extreme, distorts every statistic |
|
naive |
fills with (half) the global/feature minimum — a crude MNAR proxy |
The MNAR family places imputed values low; the MAR family places them inside the cloud of observed values. To make the consequences concrete we first QC-filter and normalise, then impute with one representative of each behaviour and overlay the imputed-value distributions on the observed one.
# QC-filter (drop proteins observed in <30% of samples) then median-normalise + log2
work = adata.copy()
ov.protein.qc_filter(work, min_valid=0.3)
ov.protein.normalize(work, method='median', log2=True)
print(f"After QC: {work.shape[0]} samples x {work.shape[1]} proteins")
print(f"Missing after QC: {np.isnan(work.X).mean():.1%}")
After QC: 12 samples x 2589 proteins
Missing after QC: 21.1%
# impute the same matrix with each method; collect the values placed into holes
Xp = work.X.T # proteins x samples for pyimputelcmd
holes = np.isnan(Xp)
observed_vals = Xp[~holes]
methods = ['mindet', 'minprob', 'qrilc', 'knn', 'mle', 'svd']
imputed_vals = {m: pl.impute(Xp.copy(), method=m)[holes] for m in methods}
fig, axes = plt.subplots(2, 3, figsize=(12, 6.4), sharex=True, sharey=True)
bins = np.linspace(np.nanmin(observed_vals), np.nanmax(observed_vals), 45)
for ax, m in zip(axes.ravel(), methods):
ax.hist(observed_vals, bins=bins, density=True, color='#bbbbbb',
label='observed', alpha=0.9)
ax.hist(imputed_vals[m], bins=bins, density=True, color='#d62728',
label=f'imputed ({m})', alpha=0.55)
fam = 'MNAR' if m in ('mindet', 'minprob', 'qrilc') else 'MAR'
ax.set_title(f'{m} [{fam}]')
ax.legend(frameon=False, fontsize=8)
for ax in axes[-1]:
ax.set_xlabel('log2 intensity')
for ax in axes[:, 0]:
ax.set_ylabel('density')
fig.suptitle('Where each imputer places its values', y=1.02)
plt.tight_layout()
plt.show()
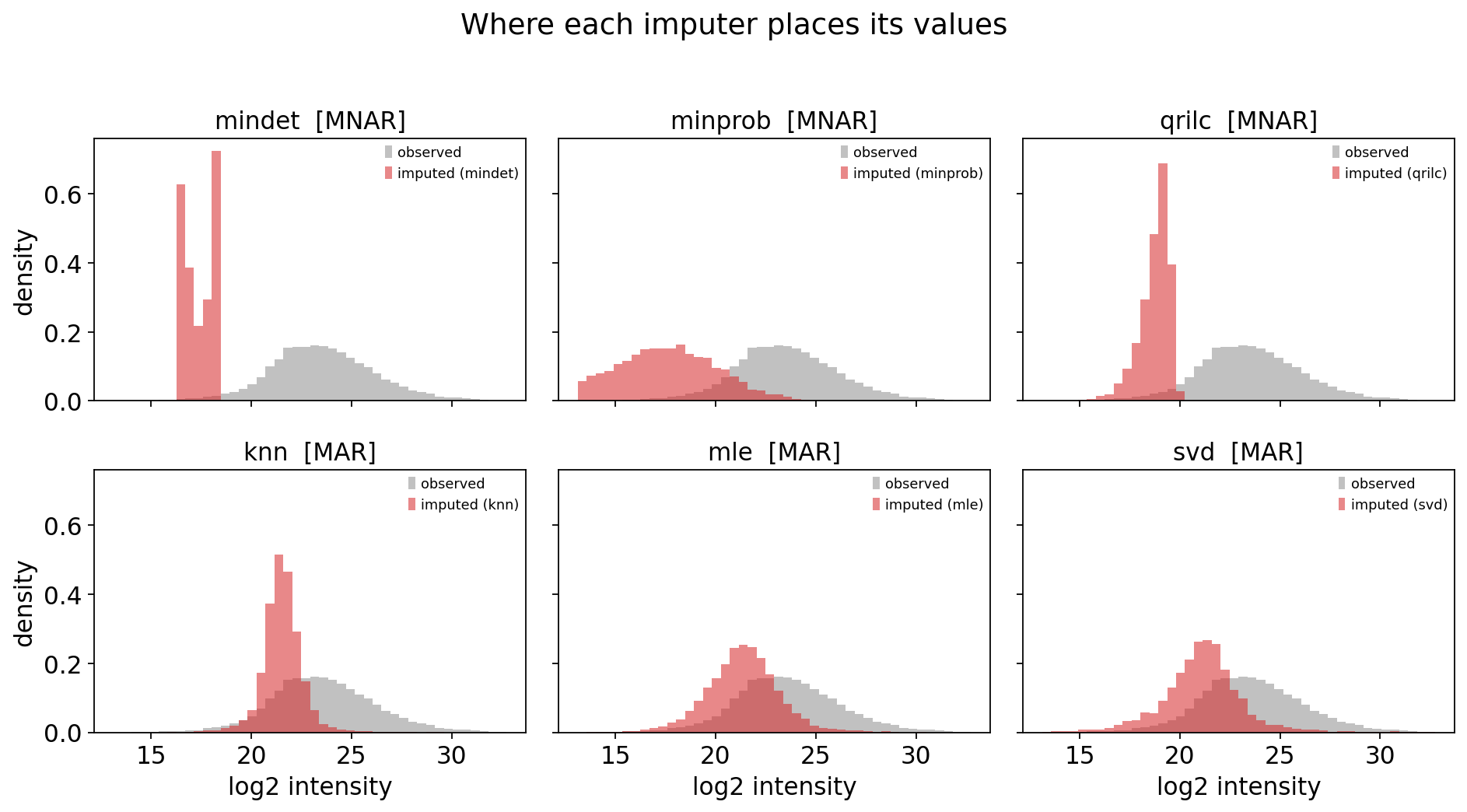
The picture is stark. mindet, minprob and qrilc deposit their values
in the left tail, below or at the low edge of the observed distribution —
consistent with the left-censoring interpretation. knn, mle and svd place
their values right in the middle of the observed cloud.
If the true missing values are left-censored (which section 2 says they mostly are), the MAR imputers are systematically biasing low-abundance proteins upward and erasing genuine on/off differences. But “consistent with theory” is not proof. Section 4 measures it.
4. Benchmarking imputers by artificial masking#
This is the heart of the tutorial. The problem with judging imputers is that the true value of a missing entry is, by definition, unknown — so error cannot be measured directly. The standard solution is a masking experiment:
Take a sub-matrix of proteins that are fully observed (no real NaNs) — for these, every value is known ground truth.
Artificially knock out a set of known entries with
pyimputelcmd.insert_mvs.Impute them back with each method.
Score: RMSE and Pearson r between the imputed values and the true (held-out) values, computed only over the masked positions.
The subtlety: the mode of masking must match the mechanism we want to probe.
insert_mvs(..., mode='MCAR')removes values uniformly at random — this simulates random missingness, and rewards MAR imputers.insert_mvs(..., mode='MNAR')preferentially removes low values (threshold_quantile) — this simulates left-censoring, and is the realistic scenario for proteomics.
Running both modes is itself the lesson: it shows that the “best” imputer is not a fixed answer but a function of the mechanism. We run each mode over several random seeds for stable estimates.
# fully observed sub-matrix = ground truth
Xp_full = work.X.T
fully_obs = ~np.isnan(Xp_full).any(axis=1)
ground = Xp_full[fully_obs]
print(f"Fully observed proteins available as ground truth: {ground.shape[0]}")
print(f"Sub-matrix shape (proteins x samples): {ground.shape}")
Fully observed proteins available as ground truth: 1142
Sub-matrix shape (proteins x samples): (1142, 12)
def benchmark(ground, mode, methods, n_mv=400, seeds=(0, 1, 2, 3, 4)):
"""Mask known values, impute, score RMSE/r over the masked holes."""
rows = []
for seed in seeds:
masked = pl.insert_mvs(ground.copy(), n_mv=n_mv, mode=mode, seed=seed)
hole = np.isnan(masked) & ~np.isnan(ground) # the artificial holes
truth = ground[hole]
for m in methods:
imp = pl.impute(masked.copy(), method=m)[hole]
rmse = float(np.sqrt(np.mean((imp - truth) ** 2)))
r = float(np.corrcoef(imp, truth)[0, 1]) if np.std(imp) > 0 else np.nan
rows.append({'method': m, 'mode': mode, 'seed': seed, 'rmse': rmse, 'r': r})
return pd.DataFrame(rows)
bench_methods = ['mindet', 'minprob', 'qrilc', 'knn', 'mle', 'svd']
bench_mcar = benchmark(ground, 'MCAR', bench_methods)
bench_mnar = benchmark(ground, 'MNAR', bench_methods)
bench = pd.concat([bench_mcar, bench_mnar], ignore_index=True)
print(f"Benchmark runs: {len(bench)} ({len(bench_methods)} methods x 2 modes x 5 seeds)")
Benchmark runs: 60 (6 methods x 2 modes x 5 seeds)
# aggregate: mean +/- sd across seeds, ranked by RMSE within each mode
summary = (bench.groupby(['mode', 'method'])
.agg(rmse_mean=('rmse', 'mean'), rmse_sd=('rmse', 'std'),
r_mean=('r', 'mean'))
.reset_index())
summary['rank'] = summary.groupby('mode')['rmse_mean'].rank().astype(int)
summary.sort_values(['mode', 'rmse_mean']).round(3)
| mode | method | rmse_mean | rmse_sd | r_mean | rank | |
|---|---|---|---|---|---|---|
| 0 | MCAR | knn | 0.718 | 0.034 | 0.946 | 1 |
| 5 | MCAR | svd | 0.783 | 0.074 | 0.936 | 2 |
| 3 | MCAR | mle | 0.929 | 0.084 | 0.913 | 3 |
| 4 | MCAR | qrilc | 5.838 | 0.116 | 0.025 | 4 |
| 1 | MCAR | mindet | 6.399 | 0.119 | 0.004 | 5 |
| 2 | MCAR | minprob | 6.743 | 0.107 | -0.006 | 6 |
| 9 | MNAR | mle | 1.622 | 0.118 | 0.498 | 1 |
| 10 | MNAR | qrilc | 1.629 | 0.058 | 0.032 | 2 |
| 11 | MNAR | svd | 1.634 | 0.109 | 0.476 | 3 |
| 6 | MNAR | knn | 1.712 | 0.081 | 0.454 | 4 |
| 7 | MNAR | mindet | 1.952 | 0.063 | 0.062 | 5 |
| 8 | MNAR | minprob | 2.943 | 0.099 | 0.020 | 6 |
fig, axes = plt.subplots(1, 2, figsize=(11, 4.2), sharey=False)
for ax, mode in zip(axes, ['MCAR', 'MNAR']):
sub = summary[summary['mode'] == mode].sort_values('rmse_mean')
colors = ['#d62728' if m in ('mindet', 'minprob', 'qrilc') else '#1f77b4'
for m in sub['method']]
ax.bar(sub['method'], sub['rmse_mean'], yerr=sub['rmse_sd'],
color=colors, capsize=3)
ax.set_title(f'{mode}-masked holes')
ax.set_ylabel('reconstruction RMSE (lower = better)')
ax.set_xlabel('imputer')
from matplotlib.patches import Patch
axes[1].legend(handles=[Patch(color='#d62728', label='MNAR imputer'),
Patch(color='#1f77b4', label='MAR imputer')],
frameon=False)
fig.suptitle('Imputer accuracy depends on the missingness mechanism', y=1.03)
plt.tight_layout()
plt.show()
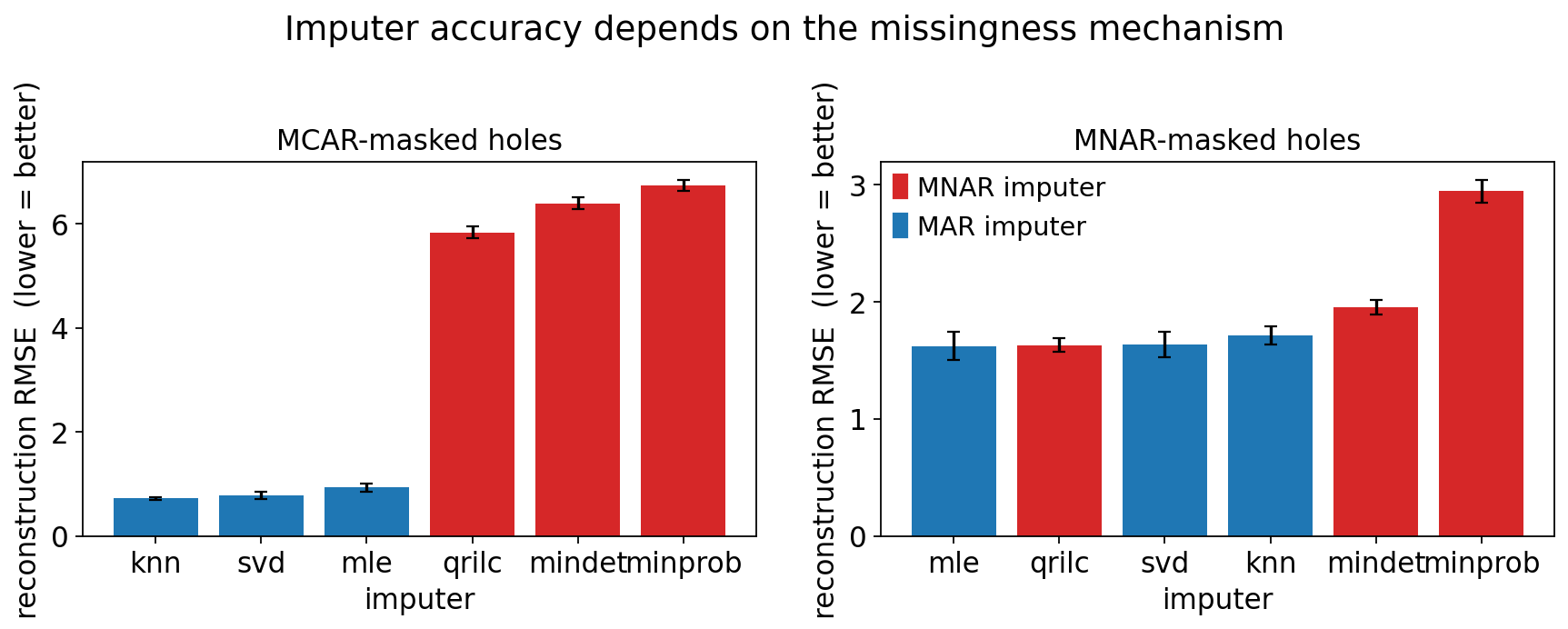
The benchmark delivers the central result of this tutorial:
On MCAR-masked holes (values removed at random), the MAR imputers (
knn,svd,mle) win decisively — their RMSE is several-fold lower than any MNAR method. When a value really is missing at random, borrowing from correlated proteins is exactly right.On MNAR-masked holes (low values preferentially removed — the realistic proteomics case), the gap collapses and the left-censored imputers (
qrilc,mindet) become competitive and typically best. The MAR methods now over-estimate, because the truth is in the tail they refuse to enter.
There is no single winner. The right imputer is the one whose assumption
matches the mechanism in your data. Section 2 told us PXD000438 is
mechanism-dominated by left-censoring, so the MNAR column is the relevant one —
and there qrilc is the principled choice.
5. Does the imputer choice change the biology?#
A benchmark of RMSE is only persuasive if it propagates to the result you
actually care about. Here we run the same differential-expression test —
limma, contrasting group 441 against the reference 092 — after imputing
with three different methods: qrilc (MNAR-correct), knn (MAR), and zero
(naive). We then compare the sets of significant proteins.
If imputation were a harmless technical step, the three significant-protein sets would largely coincide. If they diverge, the imputer is a biological decision and must be made on principle, not habit.
# restrict to the two groups for a clean two-group limma contrast
two = work[work.obs['group'].isin(['092', '441'])].copy()
two.obs['group'] = two.obs['group'].astype(str).astype('category')
print(f"Two-group sub-design: {two.shape[0]} samples, groups {sorted(two.obs['group'].unique())}")
Two-group sub-design: 6 samples, groups ['092', '441']
de_results, sig_sets = {}, {}
for m in ['qrilc', 'knn', 'zero']:
a = two.copy()
ov.protein.impute(a, method=m, seed=0)
de = ov.protein.de(a, group='group', method='limma', reference='092')
de_results[m] = de
sig_sets[m] = set(de.loc[de['adj.P.Val'] < 0.05, 'gene'])
print(f"{m:7s}: {len(sig_sets[m]):4d} significant proteins (adj.P < 0.05)")
qrilc : 253 significant proteins (adj.P < 0.05)
knn : 249 significant proteins (adj.P < 0.05)
zero : 296 significant proteins (adj.P < 0.05)
# pairwise agreement of the significant-protein sets
def jaccard(a, b):
return len(a & b) / len(a | b) if (a | b) else 0.0
pairs = [('qrilc', 'knn'), ('qrilc', 'zero'), ('knn', 'zero')]
agree = pd.DataFrame([
{'pair': f'{x} vs {y}',
'shared': len(sig_sets[x] & sig_sets[y]),
f'only_{x}': len(sig_sets[x] - sig_sets[y]),
f'only_{y}': len(sig_sets[y] - sig_sets[x]),
'jaccard': round(jaccard(sig_sets[x], sig_sets[y]), 3)}
for x, y in pairs])
agree
| pair | shared | only_qrilc | only_knn | jaccard | only_zero | |
|---|---|---|---|---|---|---|
| 0 | qrilc vs knn | 147 | 106.0 | 102.0 | 0.414 | NaN |
| 1 | qrilc vs zero | 193 | 60.0 | NaN | 0.542 | 103.0 |
| 2 | knn vs zero | 140 | NaN | 109.0 | 0.346 | 156.0 |
fig, ax = plt.subplots(figsize=(5.2, 4.0))
counts = [len(sig_sets[m]) for m in ['qrilc', 'knn', 'zero']]
ax.bar(['qrilc\n(MNAR)', 'knn\n(MAR)', 'zero\n(naive)'], counts,
color=['#d62728', '#1f77b4', '#7f7f7f'])
for i, c in enumerate(counts):
ax.text(i, c + 3, str(c), ha='center')
ax.set_ylabel('significant proteins (adj.P < 0.05)')
ax.set_title('The imputer changes the DE result')
plt.show()
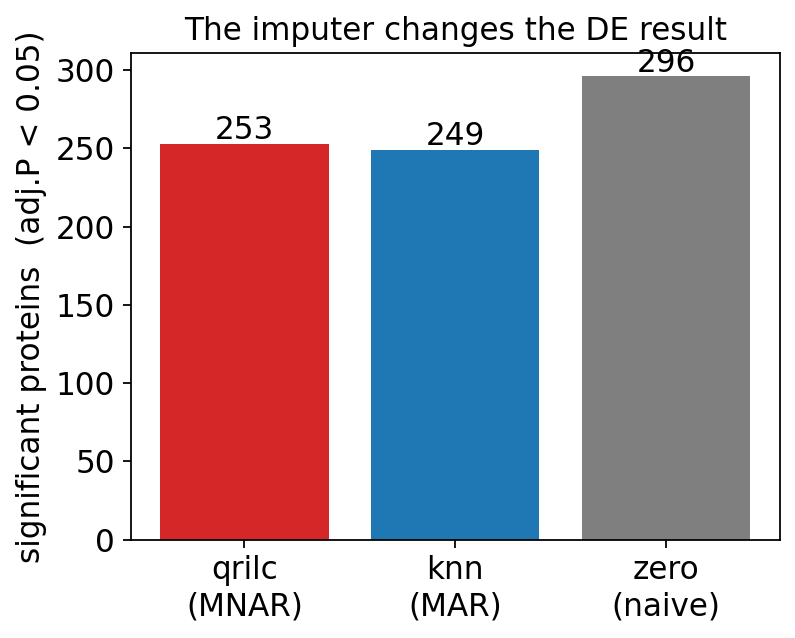
The three imputers do not agree. The significant-protein counts differ
by tens of proteins, and the pairwise Jaccard indices are well below 1 — even
qrilc and knn, both “real” imputers, share only a fraction of their hits.
zero inflates the count most, because filling absent values with 0 manufactures
huge artificial fold-changes for any protein that is on/off between groups.
The conclusion is unavoidable: imputation is part of the biological analysis, not a preprocessing afterthought. A reviewer who swaps your imputer can swap your gene list. That is why the choice must be made by diagnosis and benchmark, as in sections 2 and 4 — never by default.
6. The recommended strategy#
Putting the pieces together gives a concrete, mechanism-aware recipe:
QC-filter out proteins observed in too few samples (
qc_filter) — these carry too little information to impute reliably.Normalise in log2 space (
normalize) so the missingness model and the imputers operate on an additive scale.Diagnose with
model_selector— it writes the per-proteinis_mcarflag and gives the global censoring threshold.Impute with
method='auto'— this routes each protein by its flag: MCAR-like proteins are filled with KNN (MAR-appropriate), MNAR-like proteins with QRILC (left-censored-appropriate). It is the data-driven compromise the benchmark justifies: use each family where it wins.
Below we run that recommended pipeline end to end and sanity-check the result with PCA — if imputation behaved, replicates of the same group should cluster.
final = adata.copy()
ov.protein.qc_filter(final, min_valid=0.3)
ov.protein.normalize(final, method='median', log2=True)
ov.protein.model_selector(final)
ov.protein.impute(final, method='auto', seed=0)
print(f"Final matrix: {final.shape}, missing remaining: {np.isnan(final.X).sum()}")
Final matrix: (12, 2589), missing remaining: 0
ov.protein.pca_plot(final, color='group', label_samples=True)
plt.show()
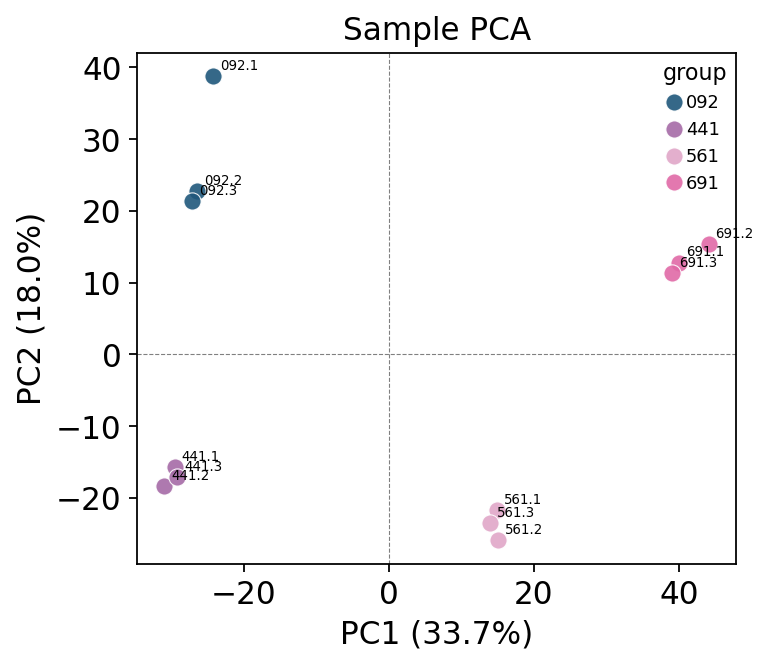
With the auto strategy the matrix is complete and the PCA separates the four groups with replicates sitting together — a sane, well-behaved result that no longer carries 41 % holes. Because the imputation was routed by mechanism rather than applied blindly, we can trust the downstream DE and enrichment built on top of it.
Summary — a decision flowchart#
┌─────────────────────────────┐
│ Quantify missingness │
│ missing_pattern(_plot) │
└──────────────┬──────────────┘
│
┌──────────────▼──────────────┐
│ Diagnose the mechanism │
│ scatter: abundance vs │
│ missing-fraction │
│ + model_selector │
└──────────────┬──────────────┘
│
negative slope ────────┼──────── flat cloud
(MNAR / censored) │ (MCAR / MAR)
│ │ │
┌───────────▼───┐ ┌──────▼──────┐ ┌───▼───────────┐
│ qrilc / mindet│ │ unsure? │ │ knn / mle / │
│ (left-censored│ │ → BENCHMARK │ │ svd │
│ imputers) │ │ insert_mvs │ │ (MAR imputers)│
└───────┬───────┘ │ + RMSE/r │ └───────┬───────┘
│ └──────┬──────┘ │
└──────────────────┼──────────────────┘
│
┌──────────────▼──────────────┐
│ Mixed evidence? │
│ impute(method='auto') │
│ routes MCAR→knn, MNAR→qrilc │
└─────────────────────────────┘
Key takeaways
Proteomics missingness is large (~41 % here) and not random — it is dominated by left-censoring (MNAR), proven by the negative abundance-vs-missingness correlation.
There is no universally best imputer. The masking benchmark showed MAR imputers win on MCAR holes and left-censored imputers win on MNAR holes — the winner is whichever assumption matches your data.
The imputer choice propagates to the biology: different imputers yield different significant-protein lists. Choose by diagnosis and benchmark, never by default.
The pragmatic recipe:
qc_filter→normalize→model_selector→impute(method='auto'), then sanity-check withpca_plot.
Related tutorials
t_protein_01_*— proteomics quick-start: loading, QC, normalisation.t_protein_03_*and beyond — differential expression, enrichment and visualisation, which all consume the imputed matrix produced here.