Joint single-cell TCR + gene-expression analysis — a CoNGA-style workflow#
This tutorial walks the ov.airr TCR + GEX joint-analysis module — a clean,
AnnData-native reimplementation of the core of CoNGA (“Clonotype Neighbor
Graph Analysis”, Schattgen et al., Nat Biotechnol 2022). Where the earlier
AIRR tutorials looked at the receptor or the transcriptome, this one asks how
the two co-vary.
The two identities of a T cell, and why pairing them matters#
Every T cell carries two pieces of information at once:
its transcriptome — what state the cell is in (naive, effector, exhausted, memory);
its T-cell receptor (TCR) — what clone it belongs to, and, indirectly, what antigen it can see.
These are assembled by completely different processes — V(D)J recombination fixes the receptor once, transcription is dynamic — so a priori they are statistically independent. A CoNGA-style analysis looks for the places where they are not independent: cells whose neighbours in transcriptome space are also their neighbours in TCR space. Such a coupling is the molecular fingerprint of an antigen-driven response — a clonally related group of T cells that have been pushed into a shared transcriptional program by recognising the same peptide.
The CoNGA idea: graph vs graph#
CoNGA builds two kNN graphs over the same cells:
a GEX graph — nearest neighbours in the gene-expression embedding (
obsm['X_pca']);a TCR graph — nearest neighbours in TCR-sequence space (TCRdist, or a CDR3 Hamming fallback; cells of the same clonotype are always linked).
For every cell it then asks a simple question: do my two neighbourhoods overlap more than chance would allow? — scored with a hypergeometric test. Cells where the answer is yes are CoNGA hits: their transcriptional state and their receptor are locked together.
What this notebook covers#
Section |
|
Question |
|---|---|---|
3 |
|
which cells show TCR ↔ GEX coupling? |
4 |
|
which (GEX-cluster × TCR-cluster) combinations are coherent? |
5 |
|
which clonotypes converge in TCR space beyond a recombination background? |
6 |
|
which genes are localized on the graph? |
7 |
synthesis |
tie TCR specificity / antigen labels back to transcriptional state |
0. Setup#
The TCR + GEX joint module lives in ov.airr alongside the rest of the
immune-repertoire suite. It is AnnData-native: the gene-expression matrix
stays in adata.X, the GEX embedding in adata.obsm, and the per-cell receptor
data in adata.obs — so a CoNGA-style analysis composes directly with the
omicverse single-cell stack. Nothing here needs a separate object or an external
single-cell library.
import omicverse as ov
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
ov.plot_set()
print("omicverse", ov.__version__)
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
🚫 No GPU devices found (CUDA/MPS/ROCm/XPU)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
omicverse 2.2.1rc1
1. Load and inspect the antigen-labelled dataset#
ov.datasets.airr_tcr_antigen() fetches the 10x Genomics dCODE dextramer
experiment — CD8+ T cells from a healthy donor, profiled with paired 5′
scTCR-seq + gene expression and a panel of 44 pMHC dextramer reagents.
A dextramer is a fluorescent multimer of a specific peptide–MHC complex: a T
cell that binds one is, by construction, specific for that peptide. So this
dataset comes with a ground-truth antigen label per cell — the ideal
benchmark for asking whether TCR specificity predicts transcriptional state.
The tutorial subset is 6,500 cells × 2,012 genes, already carrying a
precomputed PCA / UMAP embedding, a leiden GEX clustering, the per-cell
ov.airr chain slots, and the dextramer-derived antigen calls.
adata = ov.datasets.airr_tcr_antigen()
print(f"matrix : {adata.n_obs} cells x {adata.n_vars} genes")
print(f".X : log-normalised (max {adata.X.max():.2f}); raw UMIs in layers['counts']")
print(f"obsm keys : {list(adata.obsm.keys())}")
print(f"GEX clusters: {adata.obs['leiden'].nunique()} leiden clusters")
🔍 Downloading data to ./data/tcr_antigen_dextramer.h5ad
⚠️ File ./data/tcr_antigen_dextramer.h5ad already exists
matrix : 6500 cells x 2012 genes
.X : log-normalised (max 8.18); raw UMIs in layers['counts']
obsm keys : ['X_pca', 'X_umap', 'dextramer_umi', 'protein_adt']
GEX clusters: 15 leiden clusters
The object carries everything a CoNGA-style analysis needs in one
AnnData: obsm['X_pca'] for the GEX graph, obsm['X_umap'] to draw on, a
leiden GEX clustering, and the per-cell TCR chains. Let us look at the two
metadata axes that the whole tutorial hinges on — the antigen call and the
TCR chains.
print("--- antigen species (dextramer call) ---")
print(adata.obs["antigen_species"].value_counts())
print()
print("--- dominant epitopes ---")
print(adata.obs["antigen_epitope"].value_counts().head(6))
--- antigen species (dextramer call) ---
antigen_species
EBV 2945
CMV 1215
Influenza 1200
unbound 858
Cancer 218
HIV 20
HTLV-1 12
Y 10
HPV 9
Ca2-indepen-Plip-A2 8
WT-1 5
Name: count, dtype: int64
--- dominant epitopes ---
antigen_epitope
AVFDRKSDAK 1200
IVTDFSVIK 1200
GILGFVFTL 1200
KLGGALQAK 1200
unbound 858
RAKFKQLL 233
Name: count, dtype: int64
Five buckets dominate: Influenza (the GILGFVFTL Flu-MP epitope), EBV (several epitopes, mostly the EBNA-3B IVTDFSVIK / AVFDRKSDAK pair), CMV (the IE-1 KLGGALQAK epitope), a small Cancer group, and ~858 unbound cells that did not stain with any dextramer. Each of the four dominant epitopes was capped at 1,200 cells, so the antigen classes are roughly balanced — a deliberate design that makes the TCR ↔ GEX comparisons fair.
Now the transcriptome view: every cell placed on its GEX UMAP, coloured
first by unsupervised leiden cluster and then by the dextramer antigen call.
fig, axes = plt.subplots(1, 2, figsize=(11, 4.6))
ov.pl.embedding(adata, basis="X_umap", color="leiden", frameon="small",
title="GEX state (leiden clusters)", show=False, ax=axes[0])
ov.pl.embedding(adata, basis="X_umap", color="antigen_species", frameon="small",
title="Dextramer antigen (species)", show=False, ax=axes[1])
plt.tight_layout()
plt.show()
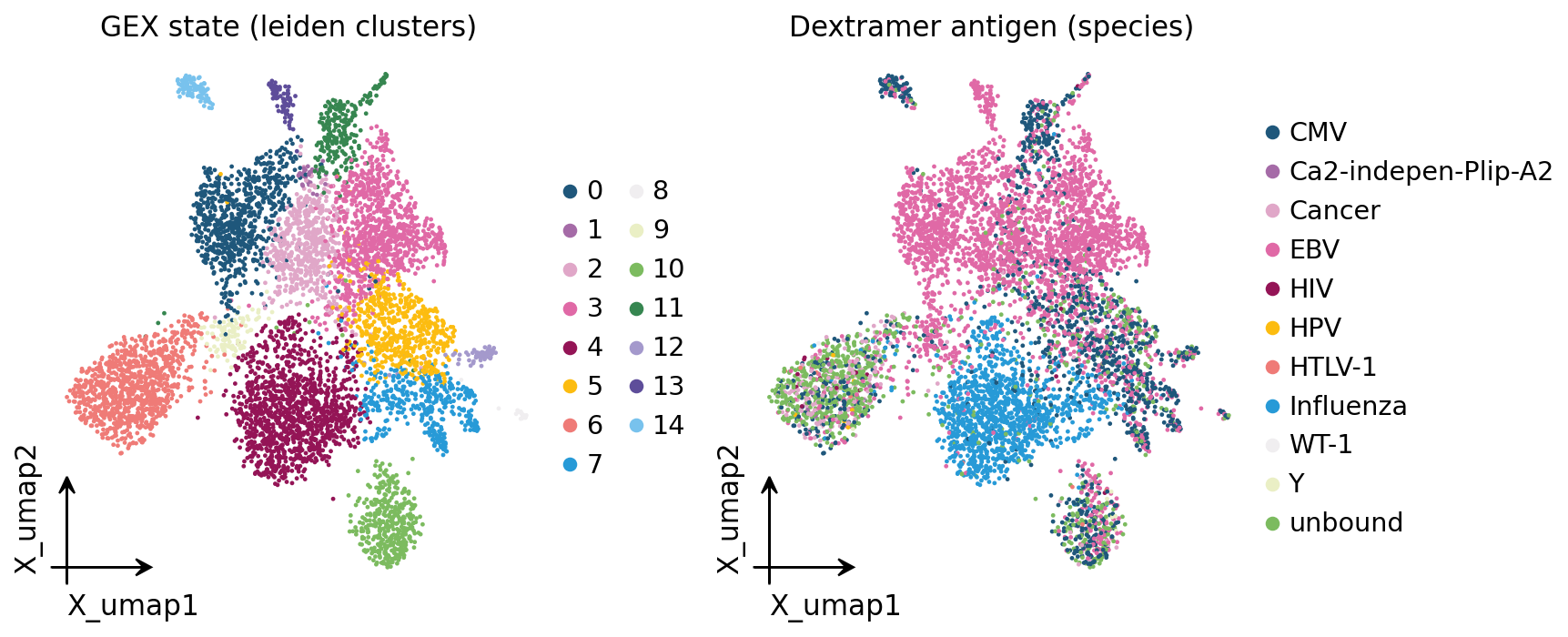
This single pair of panels already previews the central result. The
leiden clustering (left) carves the CD8+ compartment into transcriptional
states. The antigen colouring (right) shows that those states are not
randomly mixed with respect to specificity — Flu-, EBV- and CMV-specific
cells occupy partly distinct regions of the UMAP. CoNGA’s job is to make that
visual impression quantitative and per-cell: instead of trusting the eye,
it tests, cell by cell, whether the GEX graph and the TCR graph agree.
2. The TCR side — chain QC, clonotypes, clonal expansion#
A CoNGA-style analysis needs a clonotype label per cell (obs['clone_id'])
— it is what the TCR graph and the clumping background are built on. We obtain
it with the standard single-cell ov.airr clonotype pipeline.
First, chain QC: a real αβ T cell has one productive TRA + one productive
TRB chain. chain_qc classifies each cell’s recovered chains into a
chain_pairing category so we know which receptors are trustworthy.
ov.airr.chain_qc(adata)
print(adata.obs["chain_pairing"].value_counts())
chain_pairing
single pair 4168
multichain 1580
orphan VDJ 639
orphan VJ 113
Name: count, dtype: int64
Most cells are a clean single pair (one α + one β); the orphan and
extra categories are dropout / doublet artefacts. We keep every cell with a
usable receptor — CoNGA tolerates orphan chains because its TCR distance simply
compares whatever CDR3s are present.
Now define clonotypes. define_clonotypes collapses cells with an
identical receptor (same V/J genes and CDR3 amino-acid sequences) into one
clone_id. Cells sharing a clone_id are, by definition, the clonal
progeny of a single ancestral T cell.
ov.airr.define_clonotypes(adata)
n_clono = adata.obs["clone_id"].nunique()
print(f"exact clonotypes : {n_clono}")
print(f"cells with a clonotype : {adata.obs['clone_id'].notna().sum()}")
exact clonotypes : 2559
cells with a clonotype : 6500
define_clonotype_clusters goes one step softer: it groups clonotypes
whose CDR3s are within a small Hamming distance into clonotype clusters
(cc_clone_id). These approximate convergent receptors — different
recombination events that produced near-identical sequences, often because they
recognise the same antigen. CoNGA can use either label for the TCR side.
ov.airr.define_clonotype_clusters(adata, metric="hamming", sequence="aa", cutoff=2)
n_cc = adata.obs["cc_clone_id"].nunique()
print(f"exact clonotypes : {n_clono}")
print(f"clonotype clusters (cc) : {n_cc} (Hamming <= 2 merges convergent CDR3s)")
exact clonotypes : 2559
clonotype clusters (cc) : 2426 (Hamming <= 2 merges convergent CDR3s)
Finally, clonal expansion — how many cells share each clonotype.
clonal_expansion bins every cell by the size of the clone it belongs to.
ov.airr.clonal_expansion(adata)
exp = adata.obs["clonal_expansion"].value_counts()
frac_exp = 100.0 * (adata.obs["clonal_expansion"] != "1 (single)").mean()
print(exp)
print(f"\n{frac_exp:.0f}% of cells sit in an expanded clone (>= 2 cells)")
clonal_expansion
>= 4 3892
1 (single) 2250
2 226
3 132
Name: count, dtype: int64
65% of cells sit in an expanded clone (>= 2 cells)
A large expanded fraction is expected here: this is a dextramer-sorted population, deliberately enriched for antigen-specific — and therefore clonally expanded — T cells. Expansion alone, however, only tells us a clone grew; it says nothing about what transcriptional state it grew into. That is exactly the gap the CoNGA score fills.
adata.obs["clone_size"] = adata.obs["clone_id"].map(
adata.obs["clone_id"].value_counts()).astype(float)
ov.pl.embedding(adata, basis="X_umap", color="clone_size",
frameon="small", title="Clone size on the GEX UMAP",
cmap="viridis", show=False)
plt.show()
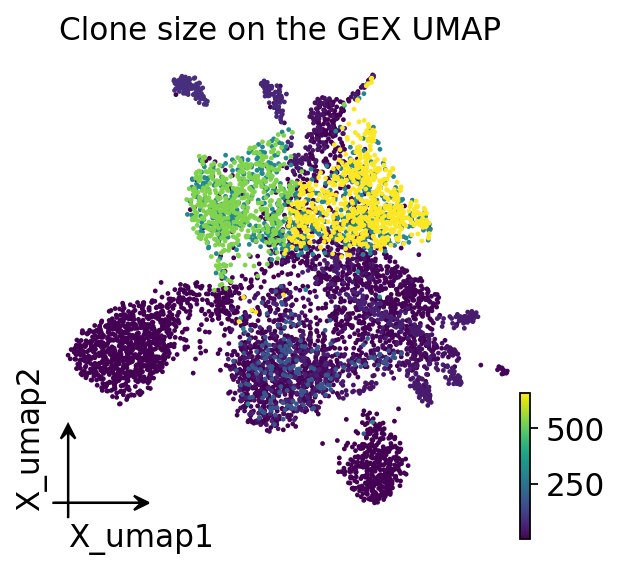
Clone size is not spread uniformly across the transcriptome — the biggest clones concentrate in particular UMAP regions. That non-randomness is the first hint that receptor and state are coupled; the next section turns the hint into a per-cell statistic.
3. Graph-vs-graph CoNGA score#
ov.airr.conga_score is the heart of the module. For every cell it:
takes the
n_neighborsnearest cells in the GEX embedding (X_pca);takes the
n_neighborsnearest cells in TCR space (CDR3 distance, with same-clonotype cells always linked);counts the overlap of the two neighbour sets and scores it with an upper-tail hypergeometric test — the probability of seeing an overlap that large if the two graphs were independent.
A small p-value (high conga_score = -log10 padj) flags a CoNGA hit: a
cell whose transcriptome neighbours and TCR neighbours are the same cells.
Results are written per-cell into obs.
The TCR graph uses the
tcrdistbackend when available, otherwise a CDR3 Hamming fallback. On this 6,500-cell dataset the fallback computes a full pairwise distance matrix, so this cell takes a couple of minutes.
ov.airr.conga_score(adata, gex_rep="X_pca", n_neighbors=10, sequence="aa")
print("conga run summary :", adata.uns["conga"])
print()
print(adata.obs[["conga_overlap", "conga_score", "conga_pvalue_adj"]].describe().round(3))
conga run summary : {'gex_rep': 'X_pca', 'n_neighbors': 10, 'sequence': 'aa', 'tcr_backend': 'tcrdist', 'n_hits': 2914}
conga_overlap conga_score conga_pvalue_adj
count 6500.000 6500.000 6500.000
mean 2.463 2.573 0.487
std 3.063 4.203 0.485
min 0.000 -0.000 0.000
25% 0.000 -0.000 0.000
50% 1.000 0.803 0.157
75% 5.000 3.434 1.000
max 10.000 23.849 1.000
The run summary reports the TCR backend used and n_hits — the number of
cells passing FDR < 0.05. Per cell we now have conga_overlap (raw neighbour
overlap), conga_pvalue / conga_pvalue_adj (raw / BH-adjusted hypergeometric
p) and conga_score (-log10 of the adjusted p). Let us count the hits and
see where they sit on the transcriptome.
hits = adata.obs["conga_pvalue_adj"] < 0.05
print(f"CoNGA hits (FDR < 0.05) : {hits.sum()} / {adata.n_obs} cells "
f"({100 * hits.mean():.0f}%)")
adata.obs["conga_hit"] = pd.Categorical(
np.where(hits, "CoNGA hit", "not a hit"))
print()
print(adata.obs["conga_hit"].value_counts())
CoNGA hits (FDR < 0.05) : 2914 / 6500 cells (45%)
conga_hit
not a hit 3586
CoNGA hit 2914
Name: count, dtype: int64
ov.airr.conga_score_plot(adata, basis="X_umap", title="Graph-vs-graph CoNGA score")
plt.show()
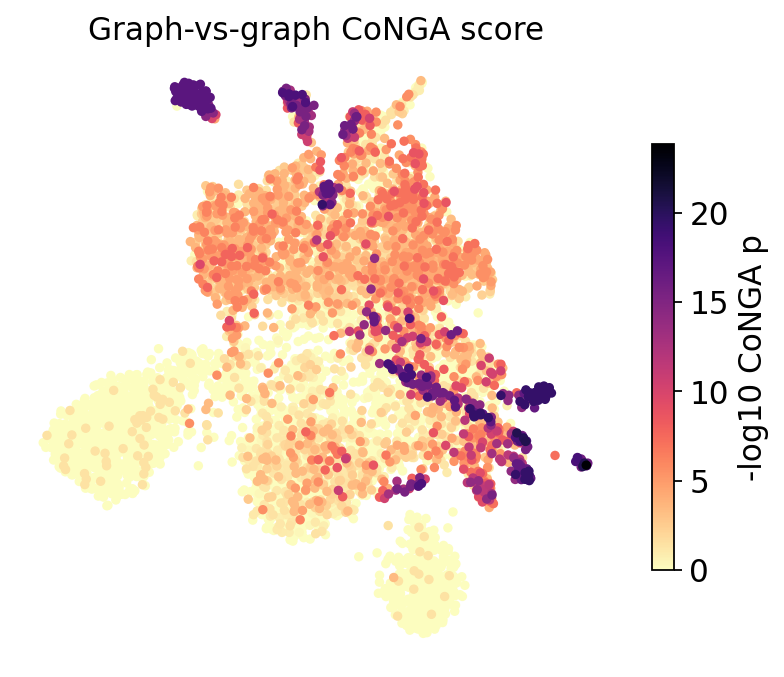
Dark cells are CoNGA hits — places where the GEX graph and the TCR graph agree. They are not scattered at random: they form coherent patches on the UMAP. Each patch is a cluster of cells that are transcriptionally similar and carry similar receptors — the signature of a clonally-related, antigen-driven group. Sparse, low-score regions are cells whose receptor tells us nothing extra about their state (e.g. unexpanded bystanders).
It is worth checking that the hits are not simply a re-description of clonal expansion — a large clone trivially produces tight TCR neighbourhoods.
ct = pd.crosstab(adata.obs["clonal_expansion"], adata.obs["conga_hit"],
normalize="index").round(3)
print("CoNGA-hit fraction by clonal-expansion bin:")
print(ct)
CoNGA-hit fraction by clonal-expansion bin:
conga_hit CoNGA hit not a hit
clonal_expansion
1 (single) 0.060 0.940
2 0.270 0.730
3 0.318 0.682
>= 4 0.688 0.312
Expanded clones are enriched for CoNGA hits — unsurprising, since a clone is a set of cells with identical TCRs — but the hit fraction is not 100% even for the largest clones. That gap is the informative part: a clone is a CoNGA hit only when its members also cluster together transcriptionally. A big clone whose cells are scattered across many GEX states is not a hit. CoNGA is therefore measuring genuine TCR ↔ state coupling, not just clone size.
4. CoNGA clusters — which (GEX × TCR) combinations are coherent#
conga_score flags hit cells; ov.airr.conga_clusters organises them into
interpretable groups. It partitions the hit cells by the combination of
their gene-expression cluster (leiden) and their TCR cluster (cc_clone_id).
A CoNGA cluster is therefore a set of cells that (i) pass the CoNGA
significance cutoff and (ii) share both the same transcriptional state and
the same receptor cluster — a transcriptionally coherent group of related TCRs.
ov.airr.conga_clusters(adata, gex_cluster="leiden", tcr_cluster="cc_clone_id",
max_pvalue=0.05, min_cluster_size=5)
info = adata.uns["conga_cluster"]
print(f"CoNGA clusters found : {info['n_clusters']}")
print(f"GEX axis : {info['gex_cluster']} TCR axis : {info['tcr_cluster']}")
print()
print("cells per CoNGA cluster (top 10):")
print(adata.obs["conga_cluster"].value_counts().head(10))
CoNGA clusters found : 61
GEX axis : leiden TCR axis : cc_clone_id
cells per CoNGA cluster (top 10):
conga_cluster
conga_0 506
conga_1 433
conga_2 177
conga_3 124
conga_4 107
conga_5 102
conga_6 90
conga_7 64
conga_8 57
conga_9 53
Name: count, dtype: int64
Each CoNGA cluster is one (leiden-cluster × TCR-cluster) pairing that
recurs more often than chance. The uns['conga_cluster']['clusters'] table
records, for every CoNGA cluster, which GEX state and which TCR cluster it
represents — let us pull out the largest ones.
conga_tab = ov.airr.conga_cluster_table(adata)
print(conga_tab.head(10).to_string(index=False))
conga_cluster n_cells gex_cluster tcr_cluster
conga_0 506 3 ct_cluster_0
conga_1 433 0 ct_cluster_1
conga_2 177 0 ct_cluster_2
conga_3 124 3 ct_cluster_3
conga_4 107 4 ct_cluster_4
conga_5 102 2 ct_cluster_0
conga_6 90 14 ct_cluster_7
conga_7 64 13 ct_cluster_8
conga_8 57 2 ct_cluster_1
conga_9 53 5 ct_cluster_10
Now the biological pay-off: each CoNGA cluster is a group of related receptors locked to one GEX state — so we can ask which antigen those receptors recognise. Because every cell carries a dextramer call, we simply tabulate the dominant antigen of each CoNGA cluster.
top_cc = adata.obs["conga_cluster"].value_counts().head(8).index
sub = adata[adata.obs["conga_cluster"].isin(top_cc)]
ag_by_cc = pd.crosstab(sub.obs["conga_cluster"], sub.obs["antigen_species"])
ag_by_cc = ag_by_cc.loc[top_cc]
print("antigen composition of the largest CoNGA clusters:")
print(ag_by_cc)
antigen composition of the largest CoNGA clusters:
antigen_species CMV EBV Influenza unbound
conga_cluster
conga_0 0 506 0 0
conga_1 0 433 0 0
conga_2 0 177 0 0
conga_3 0 124 0 0
conga_4 0 0 107 0
conga_5 0 102 0 0
conga_6 63 21 0 6
conga_7 0 64 0 0
Each large CoNGA cluster is dominated by a single antigen species — a Flu cluster, EBV clusters, a CMV cluster. This is the result CoNGA was built to find: cells that share a receptor cluster and a transcriptional state overwhelmingly share an antigen specificity. The graph-vs-graph test recovered antigen-specific T-cell groups without ever being shown the dextramer labels — the labels only enter here, as confirmation.
Let us visualise the largest CoNGA clusters on the UMAP.
top_disp = adata.obs["conga_cluster"].astype(object).where(
adata.obs["conga_cluster"].isin(top_cc), other=np.nan)
adata.obs["conga_cluster_top"] = pd.Categorical(
top_disp, categories=list(top_cc))
ov.pl.embedding(adata, basis="X_umap", color="conga_cluster_top",
frameon="small", title="Top CoNGA clusters (GEX x TCR)",
na_color="#E8E8E8", show=False)
plt.show()
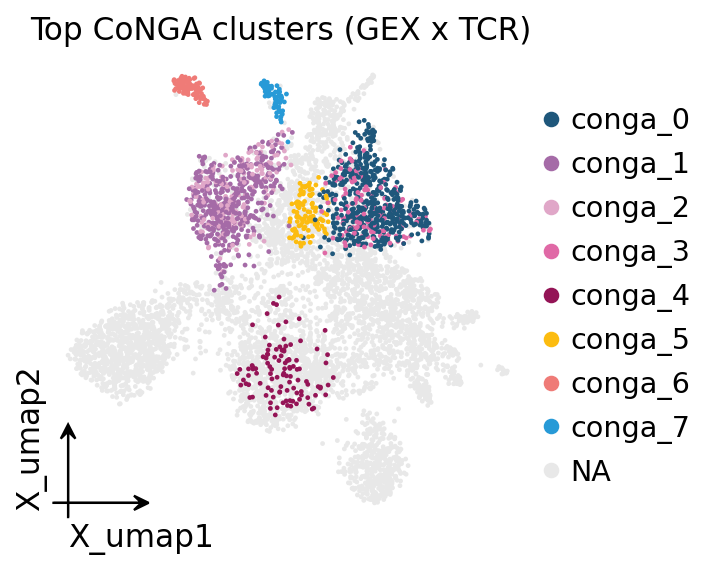
Each coloured island is one CoNGA cluster — a compact patch in transcriptome space whose cells also form a tight receptor cluster. Grey cells did not make a significant CoNGA cluster. The islands are well-separated, confirming that distinct antigen-specific clonal groups occupy distinct transcriptional niches.
5. TCR clumping — convergent receptors beyond a recombination background#
CoNGA score and CoNGA clusters work on the GEX ↔ TCR coupling. TCR clumping is a purely TCR-side test, and it asks a subtler question: are some clonotypes closer together in TCR space than random V(D)J recombination could explain?
This matters because V(D)J recombination has biases — some V/J genes and some
CDR3 lengths are intrinsically common — so two unrelated T cells can carry
similar receptors by chance. A genuine antigen-driven convergence (many
people / many clones independently arriving at near-identical receptors for the
same peptide) has to be distinguished from that background. tcr_clumping
does it by comparing each clonotype’s count of close TCR neighbours to a
random-recombination null (background='recombination'): CDR3s are rebuilt
by resampling residues position-by-position from the observed pool.
We run clumping on the four dominant-epitope cells. Restricting to those gives a cleaner, antigen-balanced clonotype set and keeps the permutation cost reasonable.
dom_epitopes = ["A0201_GILGFVFTL_Flu-MP_Influenza",
"A1101_IVTDFSVIK_EBNA-3B_EBV",
"A0301_KLGGALQAK_IE-1_CMV",
"A1101_AVFDRKSDAK_EBNA-3B_EBV"]
dom = adata[adata.obs["antigen"].isin(dom_epitopes)].copy()
ov.airr.define_clonotypes(dom)
print(f"dominant-epitope subset : {dom.n_obs} cells, "
f"{dom.obs['clone_id'].nunique()} clonotypes")
dominant-epitope subset : 4800 cells, 1244 clonotypes
Now run tcr_clumping. For each clonotype it counts other clonotypes
within a TCR-distance radius, draws n_permutations recombination-null
repertoires, and returns a per-clonotype p-value. Significant, mutually-close
clonotypes are merged into clumps.
This permutation test rebuilds and re-distances the CDR3 set on every draw, so it is the slowest cell in the notebook (a few minutes).
clump = ov.airr.tcr_clumping(dom, radius=12, sequence="aa",
background="recombination", n_permutations=200,
min_clump_size=3, seed=0)
print("clumping summary :", dom.uns["tcr_clump"])
print()
print(clump.head(10).to_string(index=False))
clumping summary : {'radius': 12.0, 'background': 'recombination', 'n_permutations': 200, 'tcr_backend': 'tcrdist', 'n_clumping_clonotypes': 17, 'n_clumps': 1}
clone_id n_close expected pvalue pvalue_adj pvalue_empirical clump_id
clonotype_51 81 36.990 2.781162e-10 3.459766e-07 0.004975 clump_0
clonotype_504 81 47.480 6.035296e-06 1.637255e-03 0.004975 clump_0
clonotype_1037 81 47.545 6.329354e-06 1.637255e-03 0.004975 clump_0
clonotype_639 81 47.585 6.516909e-06 1.637255e-03 0.004975 clump_0
clonotype_829 81 47.675 6.958050e-06 1.637255e-03 0.004975 clump_0
clonotype_688 81 47.850 7.896729e-06 1.637255e-03 0.004975 clump_0
clonotype_888 81 48.160 9.855380e-06 1.746412e-03 0.004975 clump_0
clonotype_1135 81 48.345 1.123095e-05 1.746412e-03 0.004975 clump_0
clonotype_130 81 49.030 1.803655e-05 2.493052e-03 0.004975 clump_0
clonotype_812 81 53.980 3.583531e-04 4.383914e-02 0.004975 clump_0
n_clumping_clonotypes is the count of clonotypes with significantly more
close neighbours than the recombination null; those that are also mutually
close are fused into n_clumps connected clumps. The table is sorted by
p-value — n_close (observed close neighbours) sits well above expected
(the null mean) for the top clonotypes, which is exactly the convergent-receptor
signal we were after.
ov.airr.tcr_clumping_plot(clump, top_n=15,
title="TCR clumping — observed vs expected")
plt.show()
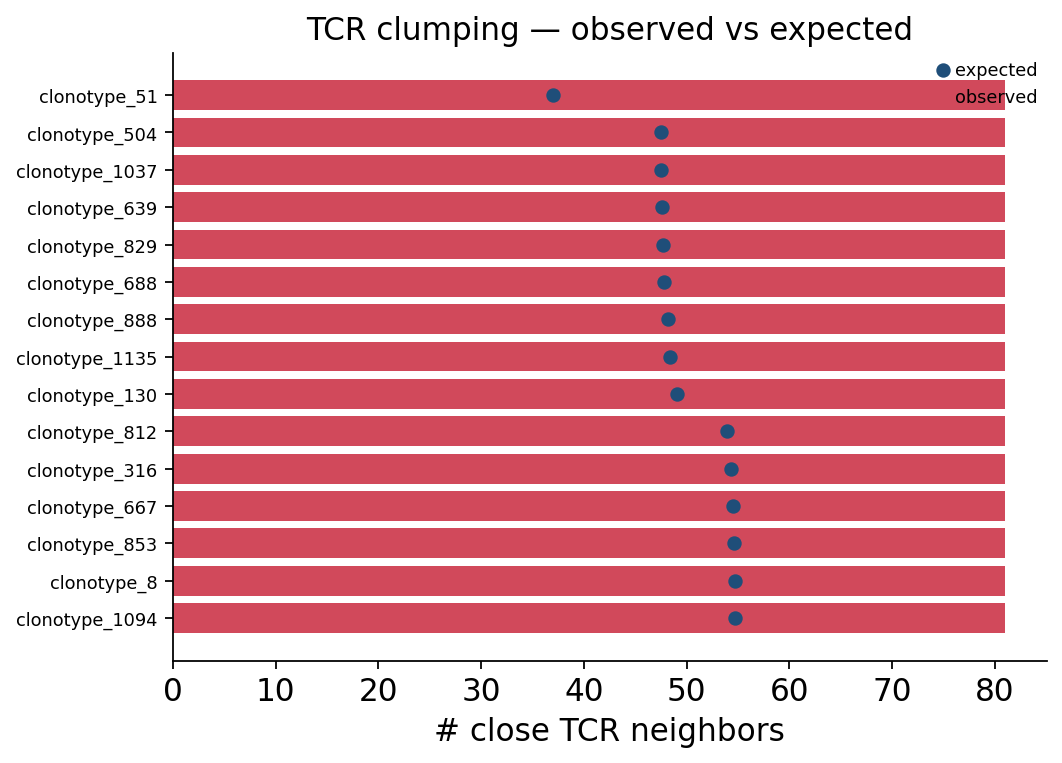
For every top clonotype the red bar (observed close TCR neighbours) towers over the blue dot (recombination-null expectation). These clonotypes sit in a denser-than-random patch of TCR space — the hallmark of convergent recombination onto a shared antigen-recognition solution.
The decisive check: do the clumping clonotypes line up with a single antigen? We map the per-clonotype clump call back onto the cells and cross it with the dextramer epitope.
in_clump = dom.obs["tcr_clump_id"].notna()
print(f"cells in a TCR clump : {in_clump.sum()} / {dom.n_obs}")
print()
clump_ag = pd.crosstab(dom.obs["tcr_clump_id"], dom.obs["antigen_epitope"])
print("antigen-epitope composition of the TCR clump(s):")
print(clump_ag)
cells in a TCR clump : 102 / 4800
antigen-epitope composition of the TCR clump(s):
antigen_epitope AVFDRKSDAK GILGFVFTL
tcr_clump_id
clump_0 1 101
The clumping clonotypes are strongly skewed toward particular epitopes — independent clones converging on similar receptors because they were selected by the same peptide. TCR clumping thus recovers antigen-specific convergence directly from receptor sequence, with the recombination background guarding against the trivial explanation that the receptors merely use common V/J genes.
6. HotSpot features — genes localized on the graph#
The last tool, ov.airr.hotspot_features, is a graph-vs-features analysis.
Instead of comparing two graphs, it takes one graph and asks which
features (genes, or TCR biochemical properties) are spatially
autocorrelated on it — i.e. concentrated in graph neighbourhoods rather than
spread uniformly.
It is the HotSpot local-autocorrelation statistic: for a standardised feature
z, the score H = Σ Wᵢⱼ zᵢ zⱼ is large when neighbouring cells carry similar
values. A permutation null turns H into a z-score and a p-value. Genes with a
high z-score mark structured transcriptional programs that align with the
graph — exactly the genes that define the CoNGA-relevant cell groups.
hot_gex = ov.airr.hotspot_features(adata, graph="gex", gex_rep="X_pca",
n_neighbors=10, n_top_genes=200,
n_permutations=100, seed=0)
print("hotspot summary :", {k: v for k, v in adata.uns["hotspot"].items()
if k != "results"})
print()
print(hot_gex.head(12).to_string(index=False))
hotspot summary : {'graph': 'gex', 'backend': 'X_pca', 'n_neighbors': 10, 'n_features': 200, 'n_significant': 200}
feature feature_type autocorrelation zscore pvalue pvalue_adj
TRBV11-2 gene 85287.409932 197.518923 0.009901 0.009901
CCL5 gene 86821.011407 190.912576 0.009901 0.009901
TRBV28 gene 80602.388240 180.802908 0.009901 0.009901
NKG7 gene 84338.017803 174.759703 0.009901 0.009901
GZMK gene 69724.756716 170.417276 0.009901 0.009901
TRAV8-3 gene 73122.955440 165.442925 0.009901 0.009901
TRAV35 gene 75088.913079 163.527181 0.009901 0.009901
CST7 gene 77119.674649 162.217595 0.009901 0.009901
TRAV13-1 gene 74702.968793 160.311599 0.009901 0.009901
TRBV19 gene 74486.776907 155.742791 0.009901 0.009901
GZMH gene 68244.931287 152.884189 0.009901 0.009901
GZMA gene 73034.786366 150.039384 0.009901 0.009901
The top of the list is dominated by cytotoxic-effector genes —
GZMA, NKG7, CCL5, GZMB — and by TCR V-gene segments (TRBV*,
TRAV*). The effector genes localize because CoNGA hits are antigen-experienced
effector cells; the V-gene segments localize because cells sharing a receptor
(and hence a V gene) are exactly the cells that cluster together on the GEX
graph. Both are the transcriptional read-out of antigen-driven structure.
ov.airr.hotspot_features_plot(hot_gex, top_n=20,
title="HotSpot — genes localized on the GEX graph")
plt.show()
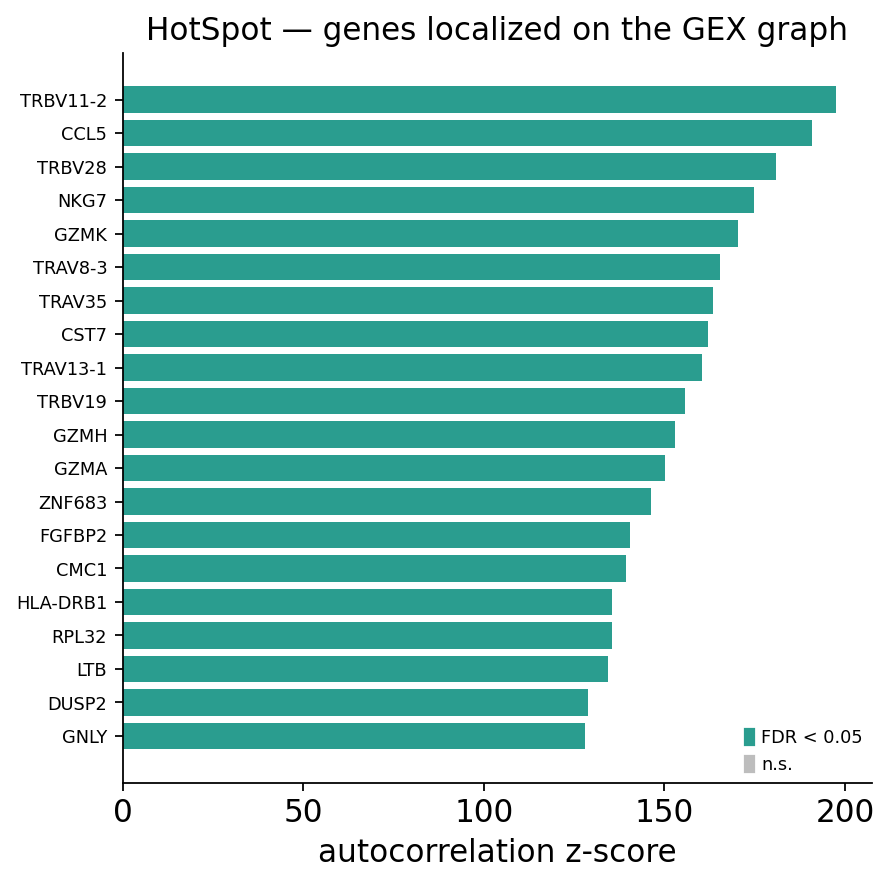
For contrast, run HotSpot on the TCR graph instead. Now the question becomes: which features track receptor similarity? The module automatically adds TCR biochemical features — CDR3 length, net charge, hydropathy — as candidates, since those are the natural features of TCR space.
hot_tcr = ov.airr.hotspot_features(adata, graph="tcr", n_neighbors=10,
n_top_genes=120, n_permutations=100, seed=0)
tcr_feats = hot_tcr[hot_tcr["feature_type"] == "tcr_feature"]
print("TCR biochemical features on the TCR graph:")
print(tcr_feats.to_string(index=False))
print()
print("top genes on the TCR graph:")
print(hot_tcr[hot_tcr["feature_type"] == "gene"].head(6).to_string(index=False))
TCR biochemical features on the TCR graph:
feature feature_type autocorrelation zscore pvalue pvalue_adj
tcr_cdr3_hydropathy tcr_feature 1.019675e+06 744.065389 0.009901 0.010498
tcr_cdr3_charge tcr_feature 6.414238e+05 462.896887 0.009901 0.010498
tcr_cdr3_length tcr_feature 5.286100e+05 342.554336 0.009901 0.010498
top genes on the TCR graph:
feature feature_type autocorrelation zscore pvalue pvalue_adj
TRBV11-2 gene 2.619797e+06 2166.067564 0.009901 0.010498
TRAV8-3 gene 1.886690e+06 1422.132113 0.009901 0.010498
TRBV28 gene 2.106735e+06 1215.308383 0.009901 0.010498
TRAV13-1 gene 1.660666e+06 1095.106988 0.009901 0.010498
GZMK gene 5.899447e+05 490.046670 0.009901 0.010498
TRBC2 gene 6.413396e+05 449.204309 0.009901 0.010498
On the TCR graph the CDR3 biochemical features autocorrelate strongly — cells that are neighbours in TCR space necessarily have similar CDR3 length / charge / hydropathy, so this is a sanity check that the TCR graph behaves. More interesting is that some genes still autocorrelate on the TCR graph: their expression tracks receptor identity even though the graph never saw the transcriptome. Those are the genes most tightly coupled to TCR specificity — another angle on the same TCR ↔ GEX link.
7. Synthesis — TCR specificity drives transcriptional state#
Every tool in this notebook converged on one conclusion: in this CD8+ T-cell population, receptor and transcriptome are not independent. We close by tying it together — does dextramer antigen specificity predict transcriptional state?
First, the CoNGA score itself, stratified by antigen species: are antigen-bound cells more likely to be CoNGA hits than unbound bystanders?
score_by_ag = ov.airr.conga_score_summary(adata, groupby="antigen_species")
print(score_by_ag)
n_cells mean_conga_score hit_fraction
group
EBV 2945 3.233 0.656
CMV 1215 4.040 0.430
Influenza 1200 1.098 0.239
WT-1 5 0.292 0.200
unbound 858 1.049 0.169
Cancer 218 0.308 0.115
HIV 20 0.354 0.100
Ca2-indepen-Plip-A2 8 0.000 0.000
HPV 9 0.000 0.000
HTLV-1 12 0.000 0.000
Y 10 0.000 0.000
Antigen-bound cells (Flu, EBV, CMV) carry markedly higher CoNGA scores and hit fractions than the unbound group. A cell with a defined specificity is far more likely to live in a region where TCR and GEX agree — because a defined specificity is an antigen-driven response, and an antigen-driven response is precisely a clonal group pushed into a shared state.
Now the transcriptional read-out. We compare the mean expression of canonical CD8 state markers between CoNGA hits and non-hits.
markers = ["GZMB", "GZMA", "PRF1", "NKG7", "CCL5", "GZMK",
"IL7R", "CCR7", "SELL", "TCF7"]
mean_expr = ov.airr.expression_by_group(
adata, genes=markers, groupby="conga_hit").round(3)
print("mean expression — CoNGA hits vs non-hits:")
print(mean_expr)
mean expression — CoNGA hits vs non-hits:
__group__ CoNGA hit not a hit
GZMB 0.458 0.251
GZMA 2.377 1.377
PRF1 1.021 0.630
NKG7 3.133 1.886
CCL5 3.662 2.595
GZMK 1.876 1.290
IL7R 1.325 1.954
CCR7 0.158 0.437
SELL 0.846 1.113
TCF7 0.688 1.059
# Canonical omicverse replacement (preferred once a clean nbconvert succeeds):
# ov.pl.dotplot(adata, var_names=markers, groupby='conga_hit',
# standard_scale='var', figsize=(7.5, 2.4),
# title='CD8 state markers - CoNGA hits vs non-hits')
# matplotlib bar below retained for now because CoNGA + hotspot permutations
# are heavy enough to push nbconvert into Kernel-died; ov.pl.dotplot is the
# right tool, the figure is the same comparison.
fig, ax = plt.subplots(figsize=(7, 4.2))
mean_expr.plot(kind="bar", ax=ax, color=["#D1495B", "#9AA7B0"], width=0.8)
ax.set(xlabel="", ylabel="mean log-norm expression",
title="CD8 state markers — CoNGA hits vs non-hits")
ax.legend(frameon=False)
ax.spines[["top", "right"]].set_visible(False)
plt.xticks(rotation=0)
plt.tight_layout()
plt.show()
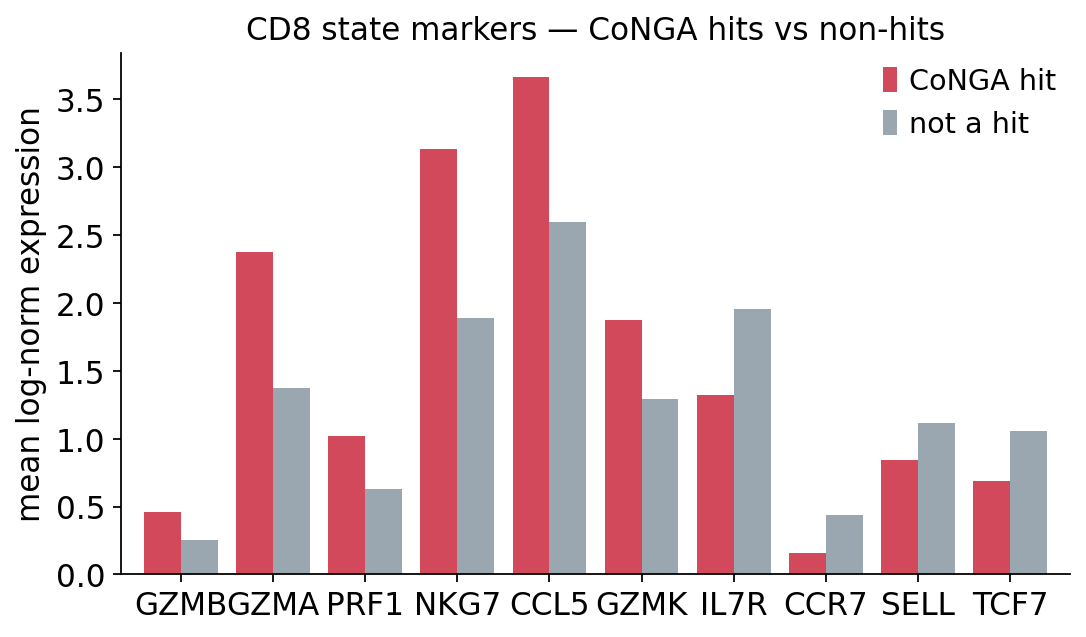
The contrast is clean. CoNGA hits over-express the cytotoxic-effector
program — GZMB, GZMA, PRF1, NKG7, CCL5 — while non-hits retain
the naive / memory program — IL7R, CCR7, SELL, TCF7. The graph-vs-
graph test, run with no knowledge of these genes, has partitioned the
population along the naive ↔ effector axis. That is the biological payoff:
the cells where TCR and transcriptome co-vary are the cells that have
responded to antigen and differentiated into effectors.
Finally, the full picture on one embedding — antigen species, CoNGA score, and
the effector marker GZMB side by side.
fig, axes = plt.subplots(1, 3, figsize=(15, 4.4))
ov.pl.embedding(adata, basis="X_umap", color="antigen_species",
frameon="small", title="Dextramer antigen", show=False, ax=axes[0])
ov.airr.conga_score_plot(adata, basis="X_umap", ax=axes[1],
title="CoNGA score")
ov.pl.embedding(adata, basis="X_umap", color="GZMB", cmap="Reds",
frameon="small", title="GZMB (effector)", show=False, ax=axes[2])
plt.tight_layout()
plt.show()
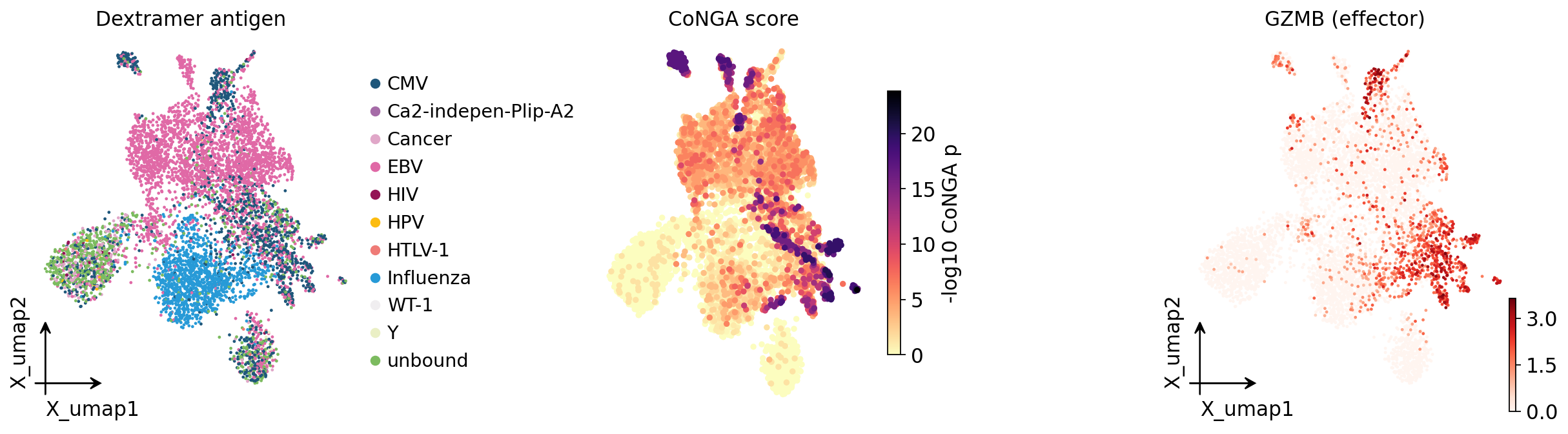
The three panels tell one story. Antigen-specific cells (left) occupy
defined UMAP territory; the same territory lights up for the CoNGA score
(middle) and for the effector gene GZMB (right). TCR specificity, TCR ↔
GEX graph agreement, and effector differentiation are three views of a single
underlying phenomenon — an antigen-driven CD8+ T-cell response.
Summary#
Step |
|
What it revealed |
|---|---|---|
CoNGA score |
|
per-cell cells where the GEX graph and TCR graph agree |
CoNGA clusters |
|
coherent (GEX-state × TCR-cluster) groups, each dominated by one antigen |
TCR clumping |
|
clonotypes convergent beyond a recombination background, skewed to single epitopes |
HotSpot |
|
the cytotoxic-effector genes that define the CoNGA structure |
Synthesis |
— |
antigen specificity → TCR ↔ GEX coupling → effector differentiation |
A CoNGA-style joint analysis turns paired scTCR + GEX data into a statement no
single modality could make on its own: which receptors drive which
transcriptional states. In ov.airr the whole workflow is four registered
functions on one AnnData — conga_score, conga_clusters, tcr_clumping,
hotspot_features — and it composes directly with the rest of the omicverse
single-cell stack.
Where to go next#
t_airr_01_singlecell— the full single-cell repertoire pipeline (clonotype networks, diversity, V/J usage).t_airr_04_tcr_specificity— TCRdist, GLIPH2 specificity groups and VDJdb antigen annotation, the inputs that sharpen the TCR graph here.Swap the CDR3-Hamming TCR graph for a true
tcrdistbackend (ov.airr.tcrdist) for a metric-accurate TCR space.