Differential expression in proteomics: a benchmark with ground truth#
When you ask “which proteins changed?”, the hard part is not computing a fold-change — it is deciding whether that fold-change is real given how few replicates a proteomics experiment usually has. Every DE test answers the same question differently: how is the per-protein variance modelled?
Method |
Variance model |
Best when |
|---|---|---|
Welch t-test |
per-protein, estimated from that protein alone |
many replicates; noisy at n = 3 |
limma |
empirical-Bayes moderated t — shrinks each protein’s variance toward one global prior |
a quick, well-calibrated default |
DEqMS |
empirical-Bayes, but the prior is conditional on peptide count |
MS data with peptide counts + few replicates |
proDA |
a probabilistic model that builds missing-not-at-random (MNAR) dropout into the test itself |
heavy, informative missingness |
Wilcoxon |
none — rank-based, non-parametric |
larger n; underpowered at n = 3 |
A peptide quantified from 20 peptides is far more reliable than one quantified from a single peptide. limma ignores that; DEqMS exploits it. This tutorial does not just run these tests — it benchmarks them on a real spike-in dataset where we know the right answer, and reports precision, recall and AUC for each.
0. Imports#
import omicverse as ov
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
print('core imports ok')
core imports ok
import pydeqms
from pydeqms.pipeline import _resolve_contrast
from sklearn.metrics import roc_auc_score
ov.plot_set()
print('omicverse', ov.__version__, '| pydeqms ok')
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
🚫 No GPU devices found (CUDA/MPS/ROCm/XPU)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
omicverse 2.2.1rc1 | pydeqms ok
1. A real benchmark with ground truth#
We use ProteomeXchange PXD000279 — the exact MaxQuant label-free dataset from the official DEqMS vignette. It is a controlled spike-in:
a constant HeLa human background (same in every sample), plus
E. coli proteins spiked in at a 3:1 ratio between two conditions (
HvsL),3 replicates per condition.
So the truth is known by construction: E. coli proteins are truly differential
(adata.var['is_spikein'] == True), human proteins are truly null. Any method that
calls many human proteins significant is producing false positives we can count.
adata = ov.datasets.protein_pxd000279()
adata
🔍 Downloading data to ./data/protein_pxd000279.h5ad
✅ Download completed
AnnData object with n_obs × n_vars = 6 × 6507
obs: 'group'
var: 'Protein_IDs', 'peptides', 'Gene_names', 'seq_coverage', 'mq_id', 'species', 'is_spikein'
uns: 'source', 'source_path'
print('samples x proteins :', adata.shape)
print('group counts :', adata.obs['group'].value_counts().to_dict())
print('species :', adata.var['species'].value_counts().to_dict())
print('true-DE (E. coli) :', int(adata.var['is_spikein'].sum()))
print('missing (NaN) frac :', round(float(np.isnan(adata.X).mean()), 3))
samples x proteins : (6, 6507)
group counts : {'H': 3, 'L': 3}
species : {'human': 4605, 'E.coli': 1902}
true-DE (E. coli) : 1902
missing (NaN) frac : 0.183
The peptide count per protein spans three orders of magnitude — exactly the spread DEqMS was built to exploit. Most proteins are quantified from only a handful of peptides.
fig, ax = plt.subplots(figsize=(5, 3))
ax.hist(adata.var['peptides'], bins=np.arange(0, 60, 2), color='#4c72b0')
ax.set_xlabel('peptides per protein'); ax.set_ylabel('# proteins')
ax.set_title('PXD000279 peptide-count distribution')
plt.show()
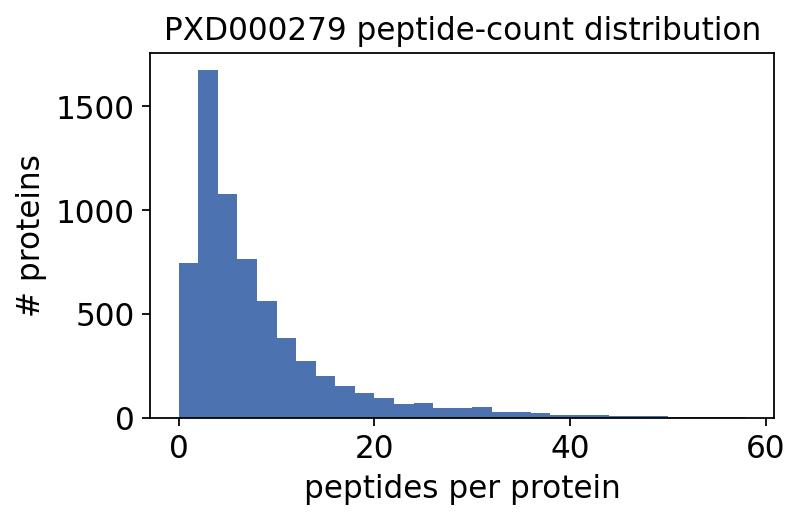
Now the standard pre-processing chain: drop proteins with too few peptides or too many missing values, median-normalise on the log2 scale, then impute the remaining missing values with QRILC (a left-censored MNAR imputer).
ov.protein.qc_filter(adata, min_peptides=2, min_valid=0.5)
ov.protein.normalize(adata, method='median', log2=True)
ov.protein.impute(adata, method='qrilc', seed=0)
print('after QC :', adata.shape, '| true-DE retained:', int(adata.var['is_spikein'].sum()))
after QC : (6, 5215) | true-DE retained: 1613
ov.protein.pca_plot(adata, color='group', label_samples=True)
plt.show()
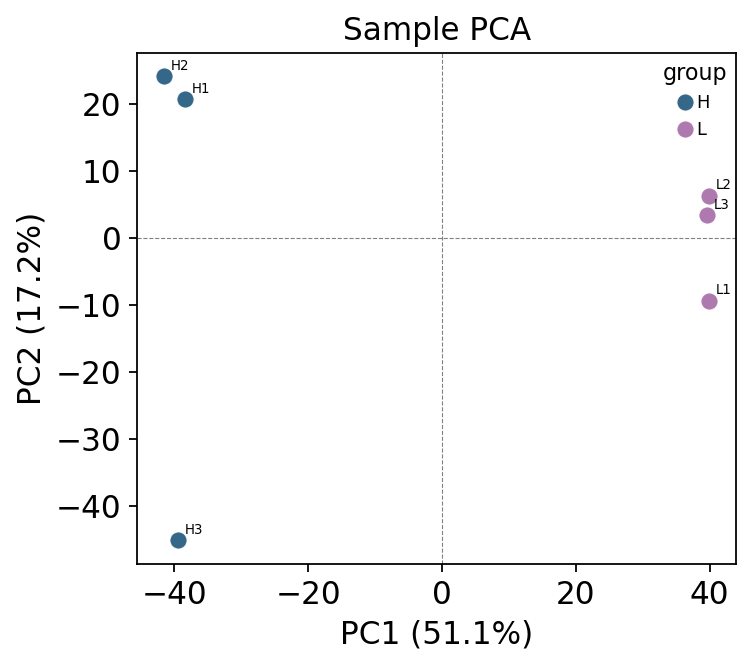
The two conditions separate cleanly on PC1 — the spike-in signal is real and strong, so any failure to detect it downstream is a failure of the test, not of the data.
2. Why peptide count matters#
Before testing, look at the data structure DEqMS depends on. We build a pydeqms
linear-model fit and plot residual variance against peptide count.
M = pd.DataFrame(adata.X.T, index=adata.var_names, columns=adata.obs_names)
design = pd.get_dummies(adata.obs['group']).astype(float)
fit = pydeqms.lm_fit(M, design)
print('design columns:', list(design.columns))
design columns: ['H', 'L']
fit, _ = _resolve_contrast(fit, np.array([-1.0, 1.0]))
counts = adata.var['peptides'].astype(float).values
pydeqms.spectra_count_ebayes(fit, count=counts)
print('eBayes with peptide-count prior fitted')
eBayes with peptide-count prior fitted
ax = pydeqms.variance_boxplot(fit, n=20)
ax.set_title('Residual variance vs peptide count')
plt.show()
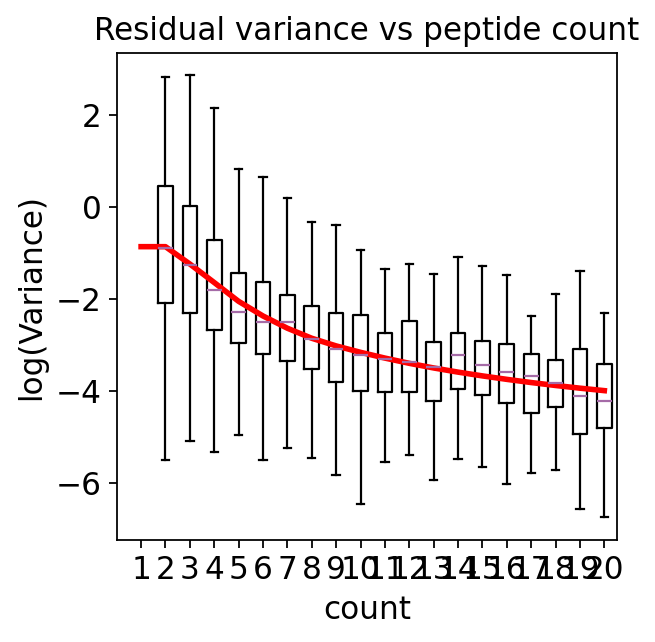
Residual variance falls steeply as peptide count rises, then plateaus. A one-peptide protein is intrinsically far noisier than a twenty-peptide protein.
limma moderates every protein toward a single global variance — it over-shrinks noisy 1-peptide proteins and under-shrinks reliable 20-peptide proteins.
DEqMS fits this curve and moderates each protein toward the prior for its own peptide count. A 1-peptide protein borrows strength from other 1-peptide proteins, not from the whole proteome. That is the entire idea, and the benchmark below shows it pays off.
3. Running five DE methods#
One call per method through ov.protein.de. reference='L' makes H the numerator, so
a positive logFC means “up in H” — and the spiked-in E. coli proteins (3:1, H:L)
should land on the positive side. count_var='peptides' is the peptide count DEqMS
needs; the other methods simply ignore it.
methods = ['welch_t', 'wilcoxon', 'limma', 'deqms', 'proda']
results = {}
for m in methods:
results[m] = ov.protein.de(adata, group='group', method=m,
reference='L', count_var='peptides')
print(f'{m:9s} -> {len(results[m])} proteins tested')
welch_t -> 5215 proteins tested
wilcoxon -> 5215 proteins tested
limma -> 5215 proteins tested
deqms -> 5215 proteins tested
proda -> 5215 proteins tested
All five methods run on the full filtered protein set (no subsampling — proDA
completes in seconds here). The DEqMS result table carries an extra count column
(the peptide count used for the conditional prior):
deqms_res = results['deqms'].sort_values('adj.P.Val')
print('DEqMS significant (adj.P < 0.05):',
int((deqms_res['adj.P.Val'] < 0.05).sum()))
deqms_res.head(8)
DEqMS significant (adj.P < 0.05): 3004
| gene | logFC | AveExpr | count | t | P.Value | adj.P.Val | |
|---|---|---|---|---|---|---|---|
| 29 | purB | 1.322213 | 29.652148 | 20.0 | 16.346227 | 1.268220e-06 | 0.000173 |
| 21 | pykF | 0.987495 | 33.593758 | 30.0 | 17.263373 | 8.882172e-07 | 0.000173 |
| 6 | polA | 1.162388 | 29.097886 | 32.0 | 20.003868 | 3.384616e-07 | 0.000173 |
| 7 | nuoG | 1.242977 | 30.311236 | 28.0 | 19.721801 | 3.715196e-07 | 0.000173 |
| 8 | rpoC | 1.159277 | 33.044972 | 69.0 | 19.507425 | 3.991340e-07 | 0.000173 |
| 39 | pta | 1.302512 | 31.867756 | 26.0 | 15.990339 | 1.463741e-06 | 0.000173 |
| 10 | valS | 1.185393 | 30.767245 | 46.0 | 18.700755 | 5.264196e-07 | 0.000173 |
| 11 | glyS | 1.301772 | 31.376737 | 42.0 | 18.514438 | 5.620962e-07 | 0.000173 |
4. Benchmarking against the spike-in truth#
This is the point of the tutorial. We have a known truth vector — E. coli proteins are the true positives, human proteins the true negatives — so we can score every method honestly. For each method at adj.P < 0.05 we count:
significant, true positives (significant ∩ E. coli), false positives (significant ∩ human),
precision = TP / significant, recall = TP / all E. coli,
ROC-AUC of the raw p-values against the truth (rank quality, threshold-free).
truth = adata.var['is_spikein'].values.astype(bool)
species = adata.var['species'].values
gene_order = adata.var_names
def bench(name, res):
res = res.set_index('gene').reindex(gene_order)
sig = (res['adj.P.Val'] < 0.05).values
tp = int((sig & truth).sum()); fp = int((sig & ~truth).sum())
n_sig = int(sig.sum())
prec = tp / n_sig if n_sig else np.nan
rec = tp / int(truth.sum())
score = -np.log10(res['P.Value'].clip(lower=1e-300).values)
auc = roc_auc_score(truth, score)
return dict(method=name, significant=n_sig, true_pos=tp, false_pos=fp,
precision=round(prec, 3), recall=round(rec, 3), auc=round(auc, 3))
bench_df = pd.DataFrame([bench(m, results[m]) for m in methods])
bench_df = bench_df.sort_values('auc', ascending=False).reset_index(drop=True)
bench_df
| method | significant | true_pos | false_pos | precision | recall | auc | |
|---|---|---|---|---|---|---|---|
| 0 | limma | 3030 | 1298 | 1732 | 0.428 | 0.805 | 0.777 |
| 1 | deqms | 3004 | 1282 | 1722 | 0.427 | 0.795 | 0.746 |
| 2 | proda | 2518 | 1150 | 1368 | 0.457 | 0.713 | 0.734 |
| 3 | welch_t | 1800 | 947 | 853 | 0.526 | 0.587 | 0.727 |
| 4 | wilcoxon | 0 | 0 | 0 | NaN | 0.000 | 0.621 |
Reading the table. A good method has high recall (it finds the real E. coli DE proteins) and high precision (it does not call human proteins). AUC summarises ranking quality independent of the 0.05 cutoff. DEqMS and proDA should top the table: DEqMS because its peptide-count-conditional prior gives noisy proteins a fair test, proDA because it models the dropout instead of imputing past it. Welch’s t-test is underpowered at n = 3; Wilcoxon, with only 20 rank permutations available, often cannot reach significance for any protein after FDR correction.
The cleanest diagnostic is the p-value histogram split by species. A well-calibrated, powerful method shows a flat uniform null for human proteins and a sharp spike near zero for E. coli proteins.
fig, axes = plt.subplots(1, 5, figsize=(18, 3), sharey=True)
bins = np.linspace(0, 1, 21)
for ax, m in zip(axes, methods):
p = results[m].set_index('gene').reindex(gene_order)['P.Value'].values
ax.hist(p[~truth], bins=bins, color='#bbbbbb', label='human (null)')
ax.hist(p[truth], bins=bins, color='#d62728', alpha=0.75, label='E. coli (true DE)')
ax.set_title(m); ax.set_xlabel('P.Value')
axes[0].set_ylabel('# proteins'); axes[0].legend(fontsize=7)
plt.tight_layout(); plt.show()
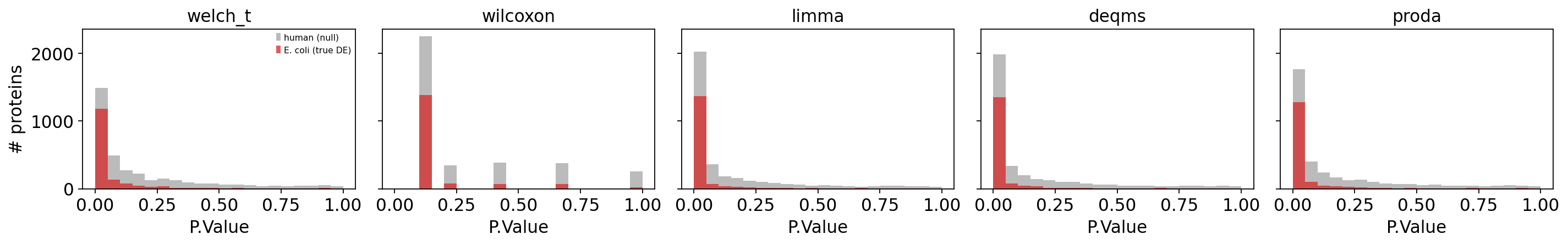
The E. coli mass piled up near P ≈ 0 is the signal; the grey human bars should be
roughly flat. A method whose human bars also spike near zero is mis-calibrated — it
would generate false discoveries on real data.
Finally, a volcano for the best-AUC method, coloured by species. The up-regulated cluster on the right is overwhelmingly E. coli — the 3:1 spike-in made visible.
best = bench_df.iloc[0]['method']
vt = results[best].set_index('gene').reindex(gene_order)
is_ecoli = truth
fig, ax = plt.subplots(figsize=(5.5, 4.5))
ax.scatter(vt['logFC'][~is_ecoli], -np.log10(vt['P.Value'][~is_ecoli]),
s=6, c='#bbbbbb', label='human (null)')
ax.scatter(vt['logFC'][is_ecoli], -np.log10(vt['P.Value'][is_ecoli]),
s=8, c='#d62728', label='E. coli (true DE)')
ax.axhline(-np.log10(0.05), ls='--', c='k', lw=0.8)
ax.set_xlabel('log2 FC (H / L)'); ax.set_ylabel('-log10 P')
ax.set_title(f'Best method: {best}'); ax.legend(fontsize=8)
plt.show()
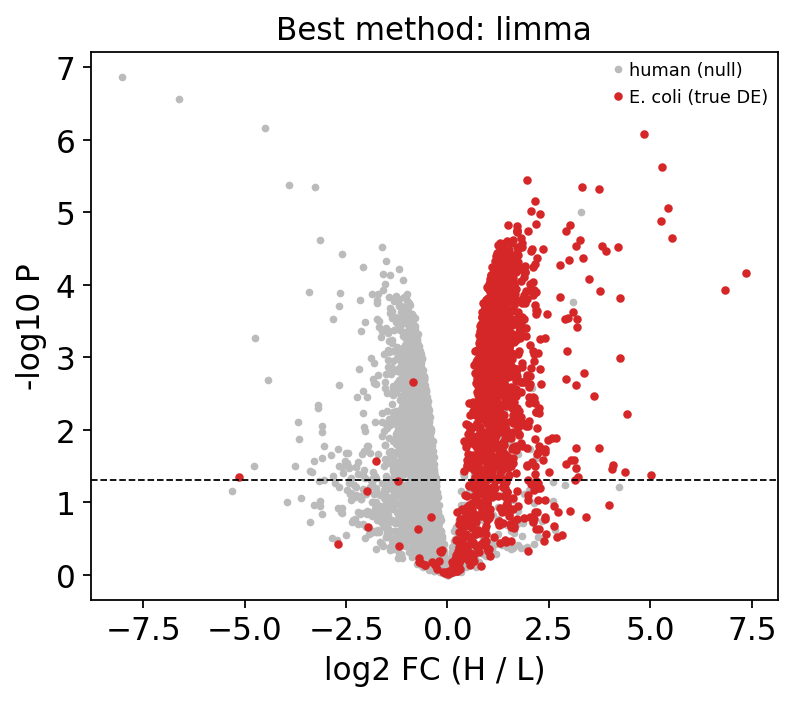
The expected spike-in ratio is log2(3) ≈ 1.58, and the red E. coli cloud centres near that value on the positive side — a real, large-scale DE result with hundreds of true differential proteins, matching the DEqMS official vignette.
5. Multi-group designs#
PXD000279 has two conditions. Real studies often have more. PXD000438 has four
groups (092 / 441 / 561 / 691), 3 replicates each. With >2 groups you first ask an
omnibus question — “does this protein differ across any group?” — with ANOVA
(parametric) or Kruskal-Wallis (rank-based).
adata4 = ov.datasets.protein_pxd000438()
ov.protein.qc_filter(adata4, min_peptides=2, min_valid=0.5)
ov.protein.normalize(adata4, method='median', log2=True)
ov.protein.impute(adata4, method='qrilc', seed=0)
print('PXD000438 after QC:', adata4.shape,
'| groups:', sorted(adata4.obs['group'].unique()))
🔍 Downloading data to ./data/protein_pxd000438.h5ad
⚠️ File ./data/protein_pxd000438.h5ad already exists
PXD000438 after QC: (12, 2194) | groups: ['092', '441', '561', '691']
anova_res = ov.protein.de(adata4, group='group', method='anova')
kruskal_res = ov.protein.de(adata4, group='group', method='kruskal')
print('ANOVA significant:', int((anova_res['adj.P.Val'] < 0.05).sum()))
print('Kruskal significant:', int((kruskal_res['adj.P.Val'] < 0.05).sum()))
anova_res.sort_values('adj.P.Val').head(6)
ANOVA significant: 1028
Kruskal significant: 0
| gene | F | AveExpr | P.Value | adj.P.Val | |
|---|---|---|---|---|---|
| 0 | P31949 | 1009.008717 | 25.336080 | 1.186731e-10 | 2.603688e-07 |
| 1 | CON__P02533 | 533.685306 | 22.847079 | 1.500758e-09 | 1.185392e-06 |
| 3 | Q9UBC9 | 486.926619 | 23.492082 | 2.161152e-09 | 1.185392e-06 |
| 2 | P07476 | 508.631952 | 22.538435 | 1.817059e-09 | 1.185392e-06 |
| 4 | P07203 | 446.600617 | 23.932371 | 3.047353e-09 | 1.337178e-06 |
| 5 | CON__P13646-1 | 355.818652 | 24.037583 | 7.512662e-09 | 2.747130e-06 |
An omnibus test tells you that a protein changes, not between which groups. For
that you specify contrasts. ov.protein.contrast_matrix turns human-readable
comparisons into the coefficient vectors a linear model consumes.
conditions = sorted(adata4.obs['group'].unique())
cmat = ov.protein.contrast_matrix(
['441-092', '561-092', '691-561'], conditions)
cmat
| 092 | 441 | 561 | 691 | |
|---|---|---|---|---|
| 441 vs 092 | -1.0 | 1.0 | 0.0 | 0.0 |
| 561 vs 092 | -1.0 | 0.0 | 1.0 | 0.0 |
| 691 vs 561 | 0.0 | 0.0 | -1.0 | 1.0 |
Each row is one comparison: +1 / -1 mark the two groups contrasted, 0 the rest.
Run the omnibus test first to control the family-wise error, then drill into the
specific contrasts that matter for your biological question.
6. Experimental design — how many replicates?#
The honest time to think about power is before the experiment. ov.protein.sample_size
uses the variance structure of pilot data (here PXD000279) to estimate how many
replicates per group you need to detect a given fold-change at 5% FDR and 80% power.
ss = ov.protein.sample_size(adata, group='group',
desired_fc=(1.5, 3.0), fdr=0.05, power=0.8)
ss.iloc[::12].reset_index(drop=True)
| desiredFC | numSample | FDR | power | CV | |
|---|---|---|---|---|---|
| 0 | 1.5 | 8 | 0.05 | 0.8 | 0.012 |
| 1 | 1.8 | 4 | 0.05 | 0.8 | 0.020 |
| 2 | 2.1 | 2 | 0.05 | 0.8 | 0.035 |
| 3 | 2.4 | 2 | 0.05 | 0.8 | 0.030 |
| 4 | 2.7 | 1 | 0.05 | 0.8 | 0.054 |
| 5 | 3.0 | 1 | 0.05 | 0.8 | 0.049 |
fig, ax = plt.subplots(figsize=(5, 3.2))
ax.plot(ss['desiredFC'], ss['numSample'], '-o', ms=3, color='#4c72b0')
ax.set_xlabel('desired fold-change'); ax.set_ylabel('replicates / group')
ax.set_title('Sample size vs effect size (FDR 5%, power 80%)')
plt.show()
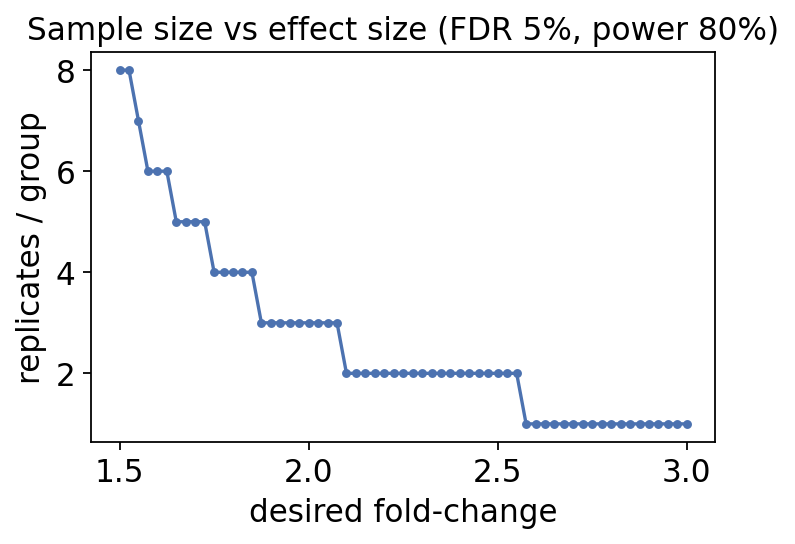
The curve is steep: subtle fold-changes demand many more replicates than large ones. A two-fold change may need only a handful of replicates, while a 1.3-fold change can need many. Run this calculation while you can still change the design — not after sequencing.
Summary — choosing a DE method#
The section-4 benchmark on real spike-in data gives a concrete decision guide:
Situation |
Use |
Why |
|---|---|---|
Peptide counts available + few replicates |
DEqMS |
conditional prior gives noisy proteins a fair, powerful test |
Heavy / informative (MNAR) missingness |
proDA |
models dropout in the test instead of imputing past it |
Quick, well-calibrated default |
limma |
global empirical-Bayes — fast and robust |
Distribution-free check, larger n |
Wilcoxon |
no parametric assumptions, but underpowered at n = 3 |
> 2 groups |
ANOVA / Kruskal then contrasts |
omnibus first, pairwise drill-down second |
Before the experiment |
|
size the study for the effect you care about |
The single lesson: at the small replicate numbers typical of proteomics, how the variance is modelled decides how many real proteins you find — DEqMS and proDA recover the spike-in truth that a plain t-test misses.