scATAC-seq preprocessing and quality control#
omicverse.epi (ov.epi) brings epigenomics into OmicVerse by wrapping the
epione toolkit. Every ov.epi function imports
epione internally and delegates, so the familiar pp / tl / pl layout you know from the
rest of OmicVerse now also covers single-cell ATAC-seq, bulk ATAC/ChIP, footprinting,
chromVAR motif activity, peak-to-gene linkage and Hi-C.
This first tutorial walks the upstream half of a single-cell ATAC-seq analysis — turning a
10x fragments.tsv.gz into a quality-controlled cell × tile matrix ready for embedding:
ingest a fragments file (
ov.epi.pp.import_fragments)compute per-cell QC metrics — TSS enrichment, fragment-size periodicity, nucleosome signal
filter low-quality cells (
ov.epi.pp.qc)build a 500 bp genome-tile count matrix and select features
a first iterative-LSI embedding to sanity-check the result
We use the public 10x PBMC 500-cell scATAC dataset (GRCh38), which ov.epi.datasets
downloads on demand — so the whole notebook is self-contained and runs on a laptop CPU.
import warnings
warnings.filterwarnings('ignore')
import numpy as np
import matplotlib.pyplot as plt
import omicverse as ov
# Confirm the epione backend is available and apply the publication plot style.
ov.epi.check_epione()
ov.epi.pl.plot_set()
print('omicverse', ov.__version__)
epione is available (version 0.0.1rc1); ov.epi is ready.
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
omicverse 2.2.1rc1
1 · Load fragments#
ov.epi.datasets.atac_pbmc500_fragments() fetches (and caches) the bgzipped, tabix-indexed
10x PBMC-500 fragments file. ov.epi.pp.import_fragments scans it into an AnnData carrying
per-cell QC counts. We pass file= so the object is written out-of-memory (anndataoom-backed) —
this is what lets the genome-wide tile matrix be added later without blowing up RAM.
The chromosome sizes / gene annotation come from ov.epi.data.hg38, a reference Genome
that lazily downloads the GRCh38 FASTA + Gencode annotation via pooch on first use.
import os
frag = ov.epi.datasets.atac_pbmc500_fragments()
print('fragments file:', frag)
genome = ov.epi.data.hg38
work = os.path.join(os.getcwd(), 'data_epi')
os.makedirs(work, exist_ok=True)
h5 = os.path.join(work, 'pbmc500.h5ad')
if os.path.exists(h5):
os.remove(h5)
adata = ov.epi.pp.import_fragments(
str(frag),
chrom_sizes=genome,
file=h5,
min_num_fragments=500,
)
adata
fragments file: /tmp/snapatac2/atac_pbmc_500.tsv.gz
└─ scanning /tmp/snapatac2/atac_pbmc_500.tsv.gz
└─ imported 646 cells (9,899,673 unique fragments)
csr_matrix float32 · 0.0% · ~0.0 MB/chunk · 0 MB disk · pbmc500.h5ad›obs3n_fragment · frac_dup · frac_mito
| name | dtype | preview |
|---|---|---|
n_fragment | int64 | 8964, 14968, 28335, … (639) |
frac_dup | float64 | 0.6372172083046663, 0.6288986958893241, 0.7626347666557765, … (645) |
frac_mito | float64 | 0.0 |
›empty–var · obsm · varm · obsp · varp · layers · raw
2 · Per-cell quality-control metrics#
Three complementary QC signals tell us which barcodes are real, high-quality cells:
TSS enrichment (
tsse) — accessibility pile-up at transcription start sites vs. flanking background. High-quality ATAC sits well above 2–4.Fragment-size distribution — open chromatin shows a clean nucleosome ladder (a sub-100 bp nucleosome-free peak, then mono-/di-nucleosome shoulders at ~200/400 bp).
Nucleosome signal — ratio of mono-nucleosome to nucleosome-free fragments per cell.
The TSS step needs gene coordinates, which we read from the bundled hg38 annotation.
genes = ov.epi.io.get_gene_annotation(genome)
print('gene annotation rows:', len(genes))
ov.epi.pp.tsse(adata, genes)
ov.epi.pp.frag_size_distr(adata)
ov.epi.pp.nucleosome_signal(adata)
# obs is a lazy on-disk element on the backed object; pull it into pandas for inspection.
obs = ov.epi.utils.obs_to_pandas(adata)
obs[['n_fragment', 'tsse', 'nucleosome_signal']].head()
gene annotation rows: 19978
└─ Computing TSS enrichment score for adata...
└─ Computing TSS enrichment score for adata...
└─ Added TSS enrichment score to adata.obs['tss_score']
└─ Created TSS enrichment score
└─ Added TSS enrichment score to tss_pileup.obs['tss_score']
└─ Returned TSS enrichment score
└─ Added TSS enrichment score to adata.obs['tsse']
└─ Computing fragment size distribution for adata...
└─ Added fragment size distribution to adata.uns['frag_size_distr']
└─ Computing nucleosome signal for adata...
└─ Added a "nucleosome_signal" column to the .obs slot of the AnnData object
└─ Created nucleosome signal
└─ Added nucleosome signal to adata.obs['nucleosome_signal']
| n_fragment | tsse | nucleosome_signal | |
|---|---|---|---|
| obs_names | |||
| AAACTGCAGACTCGGA-1 | 8964 | 27.805281 | 0.572474 |
| AAAGATGCACCTATTT-1 | 14968 | 25.742574 | 0.491104 |
| AAAGATGCAGATACAA-1 | 28335 | 39.995482 | 0.456936 |
| AAAGGGCTCGCTCTAC-1 | 28541 | 25.804671 | 0.719165 |
| AAATGAGAGTCCCGCA-1 | 7749 | 37.316490 | 0.425304 |
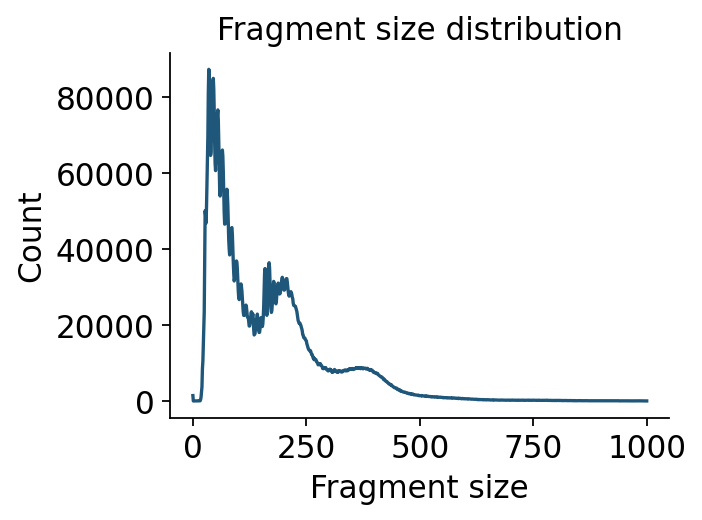
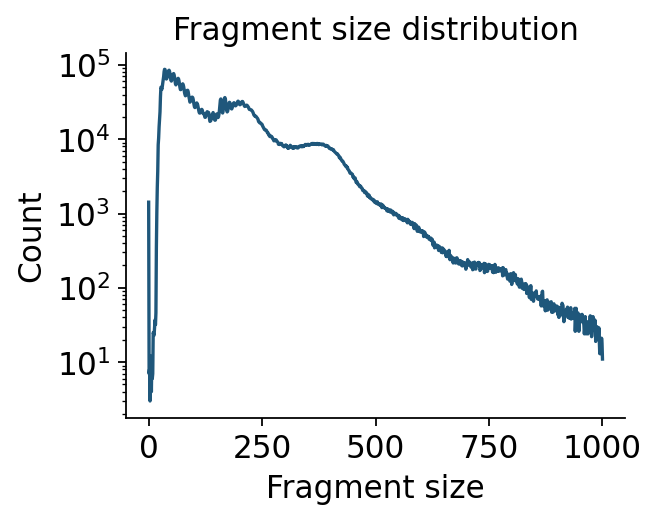
Fragment-size periodicity#
The nucleosome ladder is one of the cleanest ATAC QC signatures. ov.epi.pl.frag_size_distr
plots the distribution computed above; the log-y view makes the di-nucleosome shoulder visible.
ov.epi.pl.frag_size_distr(adata, figsize=(4, 3))
plt.show()
ov.epi.pl.frag_size_distr(adata, figsize=(4, 3), log_y=True)
plt.show()
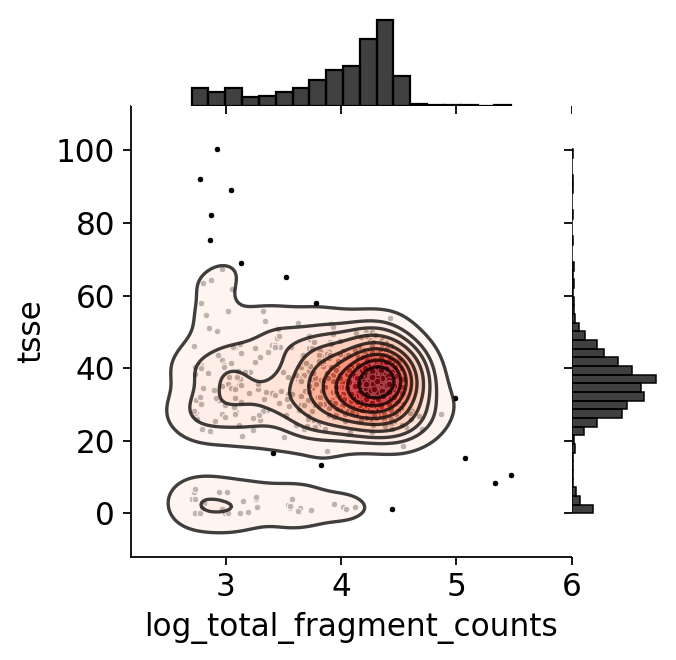
Joint QC scatter#
A log-fragment-count vs. TSS-enrichment scatter is the classic ATAC QC view: good cells sit in the upper-right (high coverage and high TSS enrichment).
# Joint QC scatter via epione's ATAC-native plotter (log-fragments vs TSSe).
obs['log_total_fragment_counts'] = np.log10(obs['n_fragment'])
ov.epi.pl.plot_joint(adata, x_col='log_total_fragment_counts', y_col='tsse',
kde_kwargs={'cut': 3})
plt.show()
3 · Filter low-quality cells#
ov.epi.pp.qc applies thresholds on all metrics at once. We keep cells with 1000–100000
fragments, TSS enrichment ≥ 2 and nucleosome signal ≤ 4.
adata = ov.epi.pp.qc(adata, tresh={
'fragment_counts_min': 1000,
'fragment_counts_max': 100000,
'TSS_score_min': 2.0,
'TSS_score_max': 100.0,
'Nucleosome_singal_max': 4.0,
})
print('cells passing QC:', adata.n_obs)
adata
└─ Performing QC...
└─ Filtering cells based on fragment counts...
└─ Filtering cells based on nucleosome signal...
└─ Filtered 78 cells
cells passing QC: 568
csr_matrix float32 · 0.0% · ~0.0 MB/chunk · 0 MB disk · pbmc500.h5ad›obs8n_fragment · frac_dup · frac_mito · tss_score · tsse · nucleosome_signal +2
| name | dtype | preview |
|---|---|---|
n_fragment | int64 | 8964, 14968, 28335, … (565) |
frac_dup | float64 | 0.6372172083046663, 0.6288986958893241, 0.7626347666557765, … (568) |
frac_mito | float64 | 0.0 |
tss_score | float64 | 27.805280528052805, 25.742574257425744, 39.995482437977635, … (560) |
tsse | float64 | 27.805280528052805, 25.742574257425744, 39.995482437977635, … (560) |
nucleosome_signal | float64 | 0.5724738675958189, 0.4911038836808877, 0.4569364161849711, … (568) |
log_total_fragment_counts | float64 | 3.9525018478630236, 4.175163774491954, 4.452323216977515, … (565) |
selected | bool | True |
›empty–var · obsm · varm · obsp · varp · layers · raw
›chain2transform pipeline
BackedArray646 × 0 · Rust (anndata-rs)_SubsetBackedArray568 × 0 · obs: 5684 · Tile matrix and feature selection#
For unbiased clustering we bin the genome into 500 bp tiles and count per-cell insertions
(add_tile_matrix), then keep the most-accessible features (select_features).
ov.epi.pp.add_tile_matrix(adata, bin_size=500)
ov.epi.pp.select_features(adata, n_features=250000)
print('tile matrix:', adata.shape)
print('features selected:', int(np.asarray(adata.var['selected']).sum()))
└─ building tile matrix: 568 cells × 6,176,584 tiles (500 bp bins, strategy=paired-insertion)
└─ tile matrix nnz=9,644,952
└─ Selected 250,000 features.
tile matrix: (568, 6176584)
features selected: 250000
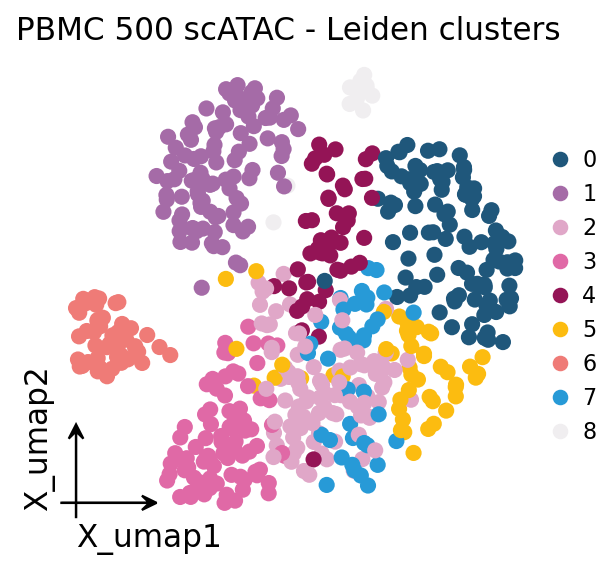
5 · A first embedding#
ArchR-style iterative LSI (ov.epi.tl.iterative_lsi) is the standard scATAC dimensionality
reduction: binarize → TF-IDF → SVD, iterated with intermediate clustering to focus on
variable features. We then build a neighbor graph, cluster and embed with UMAP — the same
pp/tl verbs as the RNA workflow.
ov.epi.tl.iterative_lsi(adata, n_components=20, iterations=2)
ov.epi.pp.neighbors(adata, use_rep='X_iterative_lsi')
ov.epi.tl.clusters(adata, method='leiden', use_rep='X_iterative_lsi', key_added='clusters')
ov.epi.tl.umap(adata)
print('embeddings:', list(adata.obsm.keys()))
└─ [iterative_lsi] Initial feature set: 500,000 / 6,176,584
└─ [iterative_lsi] Iter 1/2 | fit on 568 cells x 500,000 features
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:07)
running Leiden clustering
finished: found 7 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
└─ [iterative_lsi] -> 7 clusters; selected 25,000 variable features for next round
└─ [iterative_lsi] Iter 2/2 | fit on 568 cells x 25,000 features
└─ [iterative_lsi] Done. Stored embedding (568 x 19) in adata.obsm['X_iterative_lsi']
Computing neighbors with n_neighbors=15
Finished computing neighbors
Added to .uns['neighbors']
.obsp['distances'], distances for each pair of neighbors
.obsp['connectivities'], weighted adjacency matrix
Running Leiden clustering with leidenalg...
Finished: found 9 clusters
Added 'clusters' to adata.obs (categorical)
Computing UMAP embedding...
Finished computing UMAP
Added:
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns)
embeddings: ['X_iterative_lsi', 'X_umap']
# Plot the embedding with omicverse's standard plotter. The QC AnnData is
# anndataoom-backed, so we wrap the UMAP + cluster labels in a light in-memory
# AnnData for ov.pl.embedding.
import anndata as ad
obs = ov.epi.utils.obs_to_pandas(adata)
pa = ad.AnnData(np.zeros((adata.n_obs, 1), dtype='float32'))
pa.obs_names = list(adata.obs_names)
pa.obs['clusters'] = obs['clusters'].astype('category').values
pa.obsm['X_umap'] = np.asarray(adata.obsm['X_umap'])
ov.pl.embedding(pa, basis='X_umap', color='clusters', frameon='small',
title='PBMC 500 scATAC - Leiden clusters', show=False)
plt.show()
Summary#
Starting from a raw 10x fragments file we produced a QC-filtered, embedded scATAC dataset using
only ov.epi:
stage |
function |
|---|---|
ingest fragments |
|
TSS enrichment |
|
fragment size / nucleosome |
|
QC filter |
|
tile matrix / features |
|
embed / cluster |
|
The next tutorial picks up from a larger annotated PBMC dataset to cover clustering, cell-type annotation and gene-activity scores.