Bulk ChIP-seq upstream: FASTQ → peaks#
The earlier tutorials all started from a processed matrix. This one runs the upstream half of a
bulk ChIP-seq experiment with ov.epi.upstream (thin wrappers over bowtie2 / samtools / MACS2 /
pyBigWig): align raw reads, filter, make coverage tracks, and call peaks — then browse the result
with ov.epi.bulk.hic/ov.epi.bulk.bigwig.
Data. The Galaxy ChIP-seq training set: Tal1 transcription-factor ChIP vs. input in mouse G1E cells (GEO SRP/Zenodo record 197100), down-sampled to chr4 so the whole pipeline runs in a few minutes. The FASTQ and a regions BED are fetched from Zenodo on first run.
Tools. Needs
bowtie2,samtools,bedtools,macs2on PATH (all in the omicverse conda env). The bigwig step is purepyBigWig/pysam— no deepTools required.
Pipeline: download → build a chr4 reference → align/filter/track each replicate → merge → peaks → browse.
import os, pathlib, urllib.request, warnings
warnings.filterwarnings('ignore')
os.environ['LC_ALL'] = 'C'; os.environ['LC_CTYPE'] = 'C'
import pandas as pd
import matplotlib.pyplot as plt
import omicverse as ov
ov.epi.pl.plot_set()
print('omicverse', ov.__version__)
ov.epi.upstream.check_tools(['bowtie2', 'samtools', 'bedtools', 'macs2'])
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
omicverse 2.2.1rc1
✓ bowtie2 /scratch/users/steorra/env/omicdev/bin/bowtie2
✓ samtools /home/users/steorra/miniforge3/bin/samtools
✓ bedtools /home/users/steorra/miniforge3/bin/bedtools
✓ macs2 /scratch/users/steorra/env/omicdev/bin/macs2
{'bowtie2': '/scratch/users/steorra/env/omicdev/bin/bowtie2',
'samtools': '/home/users/steorra/miniforge3/bin/samtools',
'bedtools': '/home/users/steorra/miniforge3/bin/bedtools',
'macs2': '/scratch/users/steorra/env/omicdev/bin/macs2'}
1 · Download the demo reads#
WORK = pathlib.Path.cwd() / 'data_epi' / 'chipseq'
FASTQ_DIR = WORK / 'fastq'; REF_DIR = WORK / 'ref'; OUT = WORK / 'result'
for p in [FASTQ_DIR, REF_DIR, OUT]: p.mkdir(parents=True, exist_ok=True)
CHIP_URLS = {
'G1E_Tal1_R1.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_Tal1_R1_downsampled_SRR492444.fastqsanger/content',
'G1E_Tal1_R2.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_Tal1_R2_downsampled_SRR492445.fastqsanger/content',
'G1E_input_R1.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_input_R1_downsampled_SRR507859.fastqsanger/content',
'G1E_input_R2.fastqsanger': 'https://zenodo.org/api/records/197100/files/G1E_input_R2_downsampled_SRR507860.fastqsanger/content',
'ChIPseq_regions_of_interest_v4.bed': 'https://zenodo.org/api/records/197100/files/ChIPseq_regions_of_interest_v4.bed/content',
}
for name, url in CHIP_URLS.items():
out = FASTQ_DIR / name
if not out.exists():
print('downloading', name); urllib.request.urlretrieve(url, out)
chip_reps = [('Tal1_R1', FASTQ_DIR / 'G1E_Tal1_R1.fastqsanger'),
('Tal1_R2', FASTQ_DIR / 'G1E_Tal1_R2.fastqsanger')]
input_reps = [('Input_R1', FASTQ_DIR / 'G1E_input_R1.fastqsanger'),
('Input_R2', FASTQ_DIR / 'G1E_input_R2.fastqsanger')]
regions_bed = FASTQ_DIR / 'ChIPseq_regions_of_interest_v4.bed'
print('reads ready:', [s for s, _ in chip_reps + input_reps])
reads ready: ['Tal1_R1', 'Tal1_R2', 'Input_R1', 'Input_R2']
2 · Build a chr4 reference#
ov.epi.upstream.fetch_genome_fasta downloads the GRCm38 (mm10) FASTA (cached); we slice out chr4
and build a bowtie2 index for it with ov.epi.upstream.prepare_reference.
from pyfaidx import Fasta
MM10_FASTA = pathlib.Path(ov.epi.upstream.fetch_genome_fasta('GRCm38'))
CHR4_FASTA = REF_DIR / 'chr4.fa'; CHR4_BT2 = REF_DIR / 'chr4'
fa = Fasta(str(MM10_FASTA)); CHR4_SIZE = len(fa['chr4'])
if not CHR4_FASTA.exists():
seq = str(fa['chr4'])
with open(CHR4_FASTA, 'w') as f:
f.write('>chr4\n')
for i in range(0, len(seq), 60): f.write(seq[i:i+60] + '\n')
ref = ov.epi.upstream.prepare_reference(fasta=CHR4_FASTA, aligner='bowtie2', index_prefix=CHR4_BT2)
print('chr4 length:', CHR4_SIZE, '| index:', ref['ref_index'])
chr4 length: 156508116 | index: /scratch/users/steorra/analysis/omicverse_dev/guide-upstream-wt/docs/Tutorials-epigenetics/data_epi/chipseq/ref/chr4
3 · Align, filter and make coverage tracks#
For each replicate: bowtie2 align → mark/remove duplicates → MAPQ≥30 filter → RPKM-normalised
bigwig. ov.epi.upstream.bigwig.bam_to_bigwig is a pure pyBigWig/pysam coverage writer.
def process(reps):
runs = {}
for name, fq in reps:
d = OUT / name; d.mkdir(parents=True, exist_ok=True)
raw, filt, bw = d / f'{name}.raw.bam', d / f'{name}.filtered.bam', d / f'{name}.rpkm.bw'
for s in [raw, filt, pathlib.Path(str(filt) + '.bai'), bw]:
if s.exists(): s.unlink()
ov.epi.upstream.bowtie2.align_fastq_to_bam(
fq1=str(fq), fq2=None, out_bam=raw, ref_index=ref['ref_index'],
threads=8, extra_args=['--very-sensitive'], remove_duplicates=True)
ov.epi.upstream.samtools.filter_bam(raw, filt, mapq=30, proper_pair=False,
drop_secondary_supp=True, drop_unmapped=True, drop_mate_unmapped=False, threads=8)
ov.epi.upstream.samtools.index_bam(filt, threads=8)
ov.epi.upstream.bigwig.bam_to_bigwig(filt, bw, bin_size=25, normalize_using='RPKM', threads=8)
raw.unlink(missing_ok=True)
runs[name] = {'bam': str(filt), 'bigwig': str(bw)}
return runs
chip_runs = process(chip_reps)
input_runs = process(input_reps)
print('aligned:', list(chip_runs) + list(input_runs))
aligned: ['Tal1_R1', 'Tal1_R2', 'Input_R1', 'Input_R2']
4 · Merge replicates and call peaks#
Merge the two ChIP (and two input) BAMs, then call Tal1 peaks against the input control with
MACS2 (ov.epi.upstream.macs2.call_peaks_macs2).
merged = OUT / 'merged'; merged.mkdir(parents=True, exist_ok=True)
chip_bam = ov.epi.upstream.samtools.merge_bams(
[chip_runs['Tal1_R1']['bam'], chip_runs['Tal1_R2']['bam']], merged / 'Tal1.filtered.bam', threads=8, index=True)
input_bam = ov.epi.upstream.samtools.merge_bams(
[input_runs['Input_R1']['bam'], input_runs['Input_R2']['bam']], merged / 'Input.filtered.bam', threads=8, index=True)
chip_bw = ov.epi.upstream.bigwig.bam_to_bigwig(chip_bam, merged / 'Tal1.rpkm.bw', bin_size=25, normalize_using='RPKM', threads=8)
input_bw = ov.epi.upstream.bigwig.bam_to_bigwig(input_bam, merged / 'Input.rpkm.bw', bin_size=25, normalize_using='RPKM', threads=8)
peak_paths = ov.epi.upstream.macs2.call_peaks_macs2(
bam=chip_bam, control_bam=input_bam, out_dir=OUT / 'peaks', name='G1E_Tal1',
genome_size=str(CHR4_SIZE), format='BAM', qvalue=0.01, keep_dup='all',
call_summits=True, nomodel=False)
peaks = pd.read_csv(peak_paths['narrowPeak'], sep='\t', header=None)
print('Tal1 peaks called:', len(peaks))
peaks.head(3)
Tal1 peaks called: 26
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr4 | 5014153 | 5014190 | G1E_Tal1_peak_1 | 22 | . | 2.96644 | 6.62093 | 2.24047 | 17 |
| 1 | chr4 | 10487101 | 10487138 | G1E_Tal1_peak_2 | 40 | . | 3.95083 | 9.00442 | 4.09716 | 26 |
| 2 | chr4 | 10871782 | 10871819 | G1E_Tal1_peak_3 | 63 | . | 3.98759 | 11.40910 | 6.39778 | 17 |
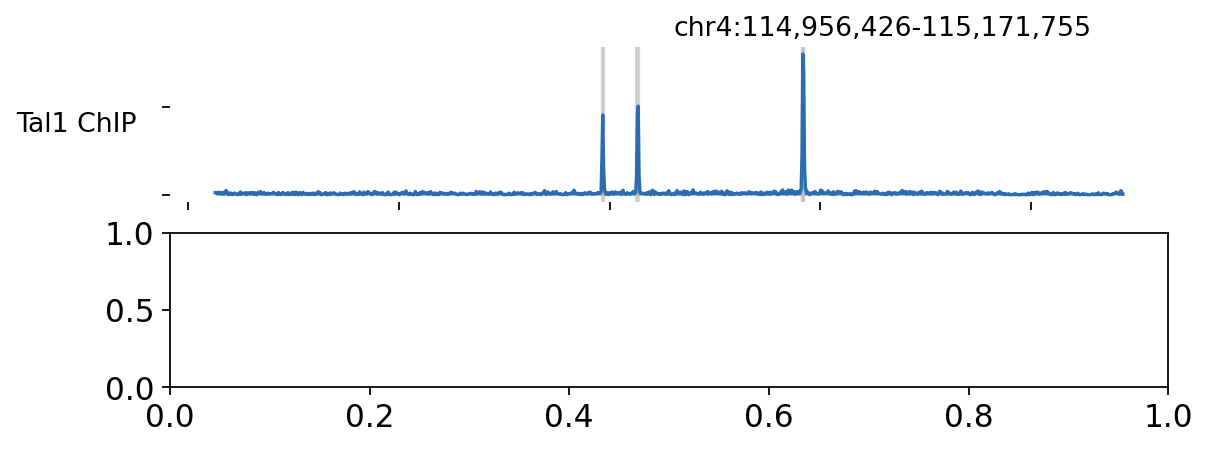
5 · Browse the Tal1 locus#
ov.epi.bulk.bigwig is a genome-browser-style track viewer. Around the Tal1 gene the ChIP track
shows a clear enrichment over input, overlapping the called peaks.
reg = pd.read_csv(regions_bed, sep='\t', header=None, names=['chrom', 'start', 'end', 'name'])
tal1 = reg[reg['name'].str.lower() == 'tal1'].iloc[0]
locus = (str(tal1['chrom']), int(tal1['start']), int(tal1['end']))
pk = peaks[(peaks[0] == locus[0]) & (peaks[1] < locus[2]) & (peaks[2] > locus[1])]
region_dict = {f'peak_{i+1}': (int(r[1]), int(r[2])) for i, (_, r) in enumerate(pk.head(8).iterrows())}
bw = ov.epi.bulk.bigwig({'Tal1 ChIP': str(merged / 'Tal1.rpkm.bw'), 'Input': str(merged / 'Input.rpkm.bw')})
bw.read()
bw.plot_track(chrom=locus[0], chromstart=locus[1], chromend=locus[2],
plot_names=['Tal1 ChIP', 'Input'], figwidth=8, figheight=2.8,
color_dict={'Tal1 ChIP': '#2B6CB0', 'Input': '#B0B0B0'},
bp_per_bin=200, region_dict=region_dict)
plt.show()
└─ Load bigWig files
├─ Loading Tal1 ChIP...
└─ Loading Input...
Summary#
stage |
function |
|---|---|
reference index |
|
align → filter → track |
|
merge replicates |
|
peak calling |
|
browse tracks |
|
This is the generic ChIP-seq / CUT&RUN upstream recipe — swap in your own FASTQ + genome and the
same ov.epi.upstream calls take you from reads to a peak set and browsable tracks.