Visualization of Bulk RNA-seq#
In this part, we will introduce the tutorial of special plot of omicverse.
import os
import warnings
warnings.filterwarnings("ignore", "Importing read_loom.*", FutureWarning)
import numpy as np
import pandas as pd
import omicverse as ov
import scanpy as sc
import matplotlib.pyplot as plt
from anndata import AnnData
os.makedirs("figures", exist_ok=True)
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
Venn plot#
In transcriptome analyses, we often have to study differential genes that are common to different groups. Here, we provide ov.pl.venn to draw venn plots to visualise differential genes.
Function: ov.pl.venn:
sets: Subgroups requiring venn plots, Dictionary format, keys no more than 4
palette: You can also re-specify the colour bar that needs to be drawn, just set
palette=['#FFFFFF','#000000'], we have preparedov.pl.red_color,ov.pl.blue_color,ov.pl.green_color,ov.pl.orange_color, by default.fontsize: the fontsize and linewidth to visualize, fontsize will be multiplied by 2
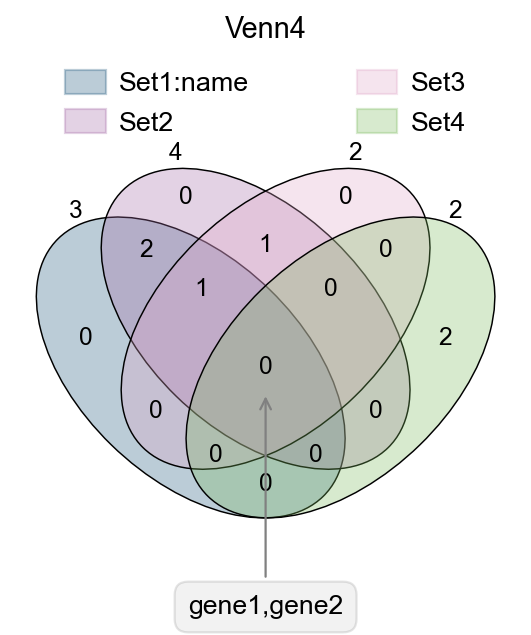
fig,ax=plt.subplots(figsize = (4,4))
#dict of sets
sets = {
'Set1:name': {1,2,3},
'Set2': {1,2,3,4},
'Set3': {3,4},
'Set4': {5,6}
}
#plot venn
ov.pl.venn(sets=sets,palette=ov.pl.sc_color,
fontsize=5.5,ax=ax,
)
#If we need to annotate genes, we can use plt.annotate for this purpose,
#we need to modify the text content, xy and xytext parameters.
plt.annotate('gene1,gene2', xy=(50,30), xytext=(0,-100),
ha='center', textcoords='offset points',
bbox=dict(boxstyle='round,pad=0.5', fc='gray', alpha=0.1),
arrowprops=dict(arrowstyle='->', color='gray'),size=12)
#Set the title
plt.title('Venn4',fontsize=13)
#save figure
fig.savefig("figures/bulk_venn4.png",dpi=300,bbox_inches = 'tight')
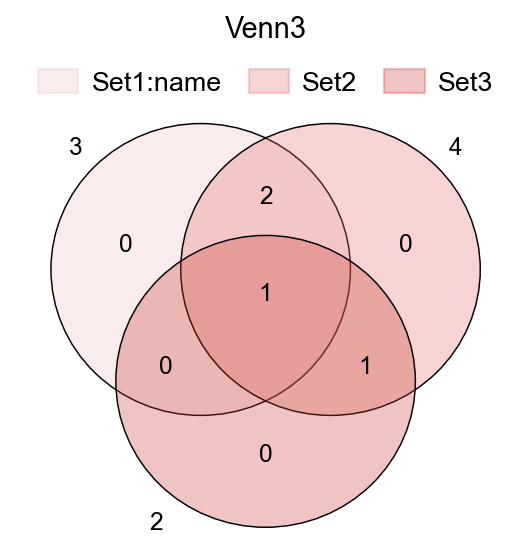
fig,ax=plt.subplots(figsize = (4,4))
#dict of sets
sets = {
'Set1:name': {1,2,3},
'Set2': {1,2,3,4},
'Set3': {3,4},
}
ov.pl.venn(sets=sets,ax=ax,fontsize=5.5,
palette=ov.pl.red_color)
plt.title('Venn3',fontsize=13)
Text(0.5, 1.0, 'Venn3')
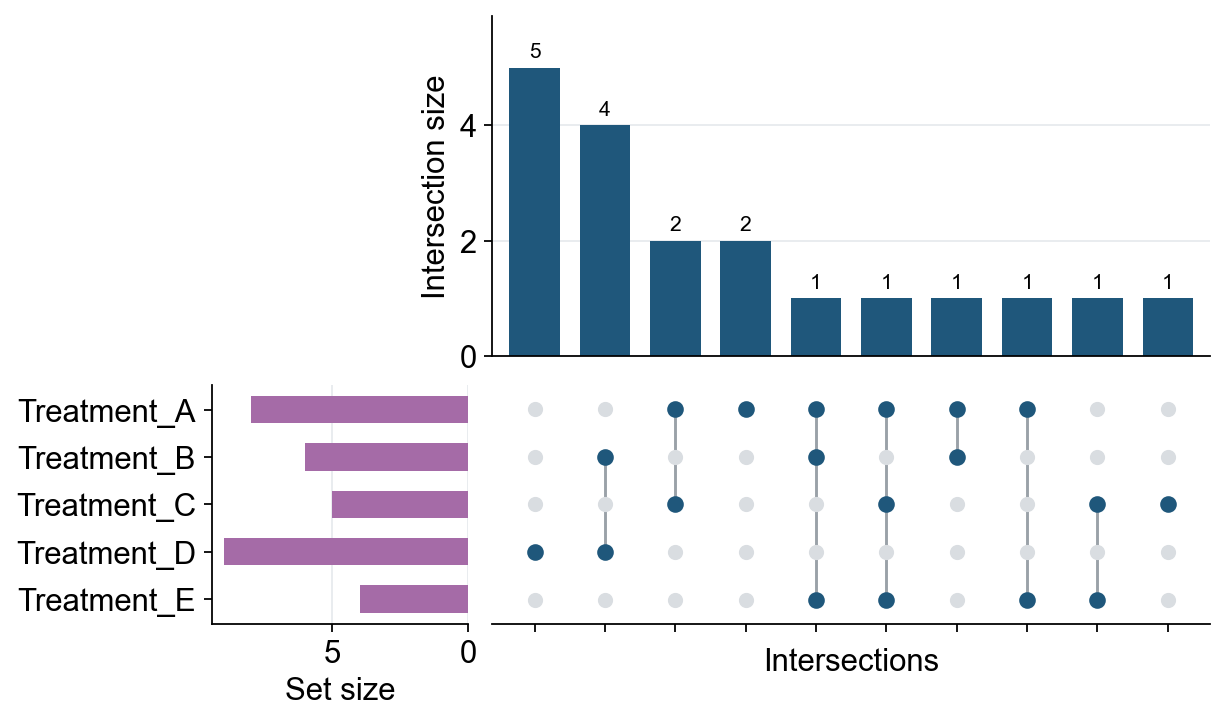
UpSet plot#
Venn plots are convenient for two to four sets. When the number of gene sets or cell-state sets is larger, an UpSet plot is usually easier to read because it represents each exclusive intersection as a bar and a dot matrix. ov.pl.upset accepts either a dictionary of Python sets or an AnnData object with boolean columns in adata.obs or adata.var.
Function: ov.pl.upset:
sets: a dictionary of sets, or anAnnDataobject.keys: forAnnData, boolean columns used as the sets. For a dictionary, optional key subset/order.axis:"obs"for cell sets or"var"for gene/feature sets when usingAnnData.top_n: maximum number of intersections to display.min_size: minimum exclusive intersection size to keep.style: passov.style,True, or a dictionary ofov.stylekeyword arguments to apply OmicVerse plotting style before drawing.
# Multiple gene sets can be displayed directly as an UpSet plot.
gene_sets = {
"Treatment_A": {"IL7R", "CCR7", "LTB", "SELL", "TCF7", "MAL", "CD3D", "CD3E"},
"Treatment_B": {"CCR7", "LTB", "NKG7", "GZMB", "PRF1", "IFNG"},
"Treatment_C": {"IL7R", "TCF7", "MAL", "LEF1", "GZMK"},
"Treatment_D": {"NKG7", "GZMB", "PRF1", "GNLY", "IFNG", "CTSW", "KLRD1", "KLRF1", "CST7"},
"Treatment_E": {"IL7R", "SELL", "LEF1", "CCR7"},
}
fig, axes = ov.pl.upset(gene_sets, top_n=15, min_size=1, figsize=(8, 5), style=ov.style)
fig.savefig("figures/bulk_upset_gene_sets.png", dpi=300, bbox_inches="tight")
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ Apple Silicon MPS detected
• [MPS] Apple Silicon GPU - Metal Performance Shaders available
✅ plot_set complete.
# AnnData input uses boolean obs/var columns as set membership indicators.
rng = np.random.default_rng(42)
adata_upset = AnnData(
np.ones((80, 5)),
obs=pd.DataFrame(index=[f"cell_{i}" for i in range(80)]),
var=pd.DataFrame(index=[f"gene_{i}" for i in range(5)]),
)
adata_upset.obs["T_cell"] = rng.random(80) < 0.55
adata_upset.obs["Activated"] = rng.random(80) < 0.42
adata_upset.obs["Cycling"] = rng.random(80) < 0.25
adata_upset.obs["IFN_response"] = rng.random(80) < 0.30
adata_upset.obs["Cytotoxic"] = rng.random(80) < 0.28
adata_upset.obs["Stress"] = rng.random(80) < 0.22
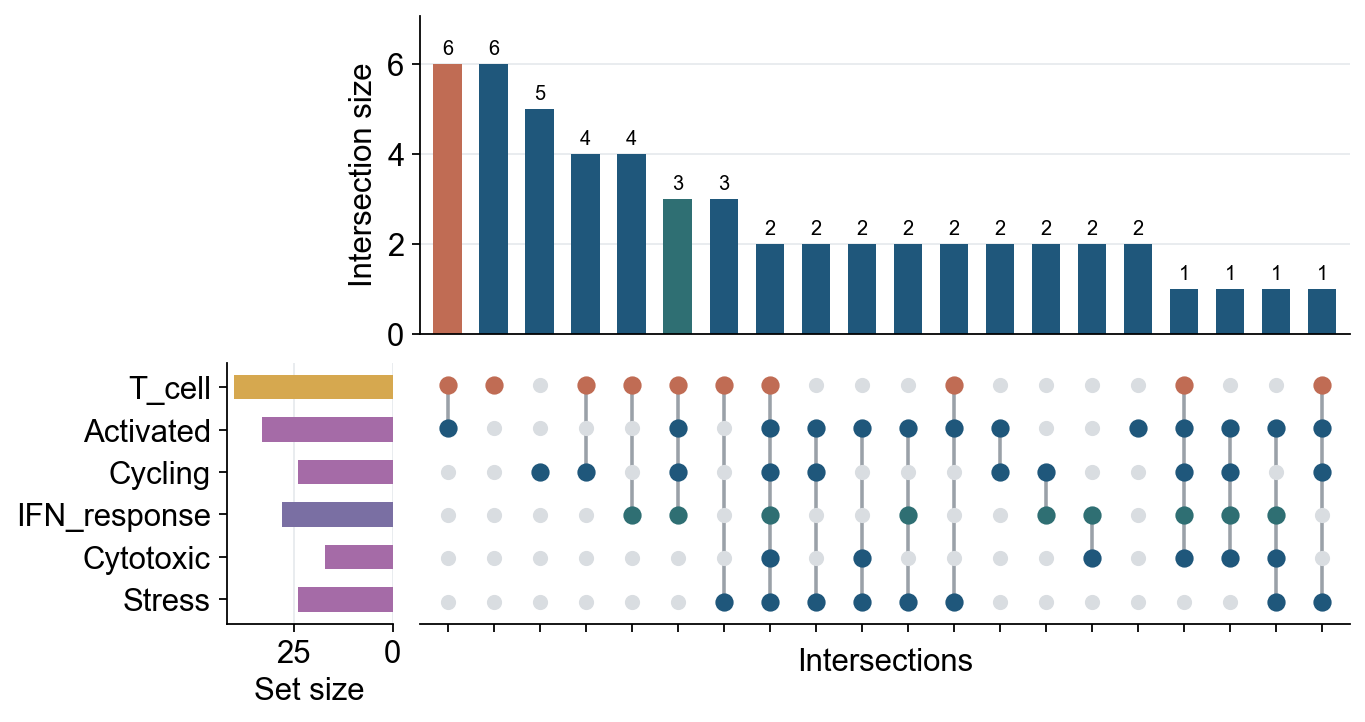
keys = ["T_cell", "Activated", "Cycling", "IFN_response", "Cytotoxic", "Stress"]
fig, axes = ov.pl.upset(
adata_upset,
keys=keys,
axis="obs",
top_n=20,
figsize=(9, 5),
intersection_color={
"T_cell&Activated": "#C06C54",
"T_cell&Activated&Cycling&IFN_response": "#2F6F73",
},
set_size_color={"T_cell": "#D6A84F", "IFN_response": "#7A6FA3"},
matrix_color={"T_cell": "#C06C54", "IFN_response": "#2F6F73"},
bar_width=0.62,
dot_size=52,
line_width=1.6,
count_fontsize=9,
height_ratios=(0.55, 0.45),
)
fig.savefig("figures/bulk_upset_adata_obs.png", dpi=300, bbox_inches="tight")
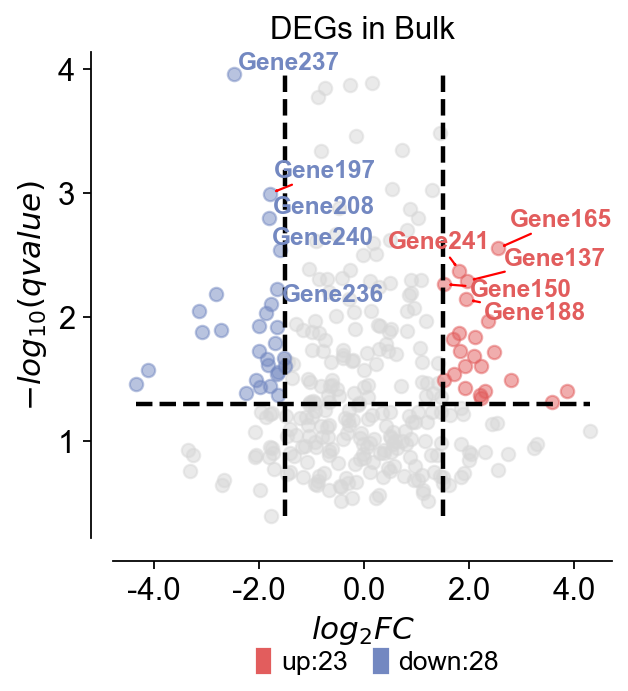
Volcano plot#
For differentially expressed genes, we tend to visualise them only with volcano plots. Here, we present a method for mapping volcanoes using Python ov.pl.volcano.
Function: ov.pl.venn:
main argument
result: the DEGs result
pval_name: the names of the columns whose vertical coordinates need to be plotted, stored in result.columns. In Bulk RNA-seq experiments, we usually set this to qvalue.
fc_name: The names of the columns for which you need to plot the horizontal coordinates, stored in result.columns. In Bulk RNA-seq experiments, we typically set this to log2FC.
fc_max: We need to set the threshold for the difference foldchange
fc_min: We need to set the threshold for the difference foldchange
pval_threshold: We need to set the threshold for the qvalue
pval_max: We also need to set boundary values so that the data is not too large to affect the visualisation
FC_max: We also need to set boundary values so that the data is not too large to affect the visualisation
plot argument
figsize: The size of the generated figure, by default (4,4).
title: The title of the plot, by default ‘’.
titlefont: A dictionary of font properties for the plot title, by default {‘weight’:’normal’,’size’:14,}.
up_color: The color of the up-regulated genes in the plot, by default ‘#e25d5d’.
down_color: The color of the down-regulated genes in the plot, by default ‘#7388c1’.
normal_color: The color of the non-significant genes in the plot, by default ‘#d7d7d7’.
legend_bbox: A tuple containing the coordinates of the legend’s bounding box, by default (0.8, -0.2).
legend_ncol: The number of columns in the legend, by default 2.
legend_fontsize: The font size of the legend, by default 12.
plot_genes: A list of genes to be plotted on the volcano plot, by default None.
plot_genes_num: The number of genes to be plotted on the volcano plot, by default 10.
plot_genes_fontsize: The font size of the genes to be plotted on the volcano plot, by default 10.
ticks_fontsize: The font size of the ticks, by default 12.
rng = np.random.default_rng(0)
genes = [f"Gene{i}" for i in range(1, 301)]
result = pd.DataFrame({
"log2FoldChange": rng.normal(0, 1.4, len(genes)),
"qvalue": np.clip(rng.beta(0.7, 7, len(genes)), 1e-6, 1),
}, index=genes)
result["sig"] = "normal"
result.loc[(result["qvalue"] < 0.05) & (result["log2FoldChange"] > 1.5), "sig"] = "up"
result.loc[(result["qvalue"] < 0.05) & (result["log2FoldChange"] < -1.5), "sig"] = "down"
result.head()
log2FoldChange qvalue sig
Gene1 0.176022 0.146827 normal
Gene2 -0.184947 0.040901 normal
Gene3 0.896592 0.005051 normal
Gene4 0.146860 0.061055 normal
Gene5 -0.749937 0.015369 normal
ov.pl.volcano(result,pval_name='qvalue',fc_name='log2FoldChange',
pval_threshold=0.05,fc_max=1.5,fc_min=-1.5,
pval_max=10,FC_max=10,
figsize=(4,4),title='DEGs in Bulk',titlefont={'weight':'normal','size':14,},
up_color='#e25d5d',down_color='#7388c1',normal_color='#d7d7d7',
up_fontcolor='#e25d5d',down_fontcolor='#7388c1',normal_fontcolor='#d7d7d7',
legend_bbox=(0.8, -0.2),legend_ncol=2,legend_fontsize=12,
plot_genes=None,plot_genes_num=10,plot_genes_fontsize=11,
ticks_fontsize=12,)
🌋 Volcano Plot Analysis:
Total genes: 300
↗️ Upregulated genes: 23
↘️ Downregulated genes: 28
➡️ Non-significant genes: 249
🎯 Total significant genes: 51
log2FoldChange range: -4.35 to 4.29
qvalue range: 1.10e-04 to 4.00e-01
⚙️ Current Function Parameters:
Data columns: pval_name='qvalue', fc_name='log2FoldChange'
Thresholds: pval_threshold=0.05, fc_max=1.5, fc_min=-1.5
Plot size: figsize=(4, 4)
Gene labels: plot_genes_num=10, plot_genes_fontsize=11
Custom genes: None (auto-select top genes)
💡 Parameter Optimization Suggestions:
✅ Current parameters are optimal for your data!
────────────────────────────────────────────────────────────
<Axes: title={'center': 'DEGs in Bulk'}, xlabel='$log_{2}FC$', ylabel='$-log_{10}(qvalue)$'>
Box plot#
For differentially expressed genes in different groups, we sometimes need to compare the differences between different groups, and this is when we need to use box-and-line plots to do the comparison
Function: ov.pl.boxplot:
data: the data to visualize the boxplt example could be found in
seaborn.load_dataset("tips")x_value, y_value, hue: Inputs for plotting long-form data. See examples for interpretation.
figsize: The size of the generated figure, by default (4,4).
fontsize: The font size of the tick and labels, by default 12.
title: The title of the plot, by default ‘’.
Function: ov.pl.add_palue:
ax: the axes of bardotplot
line_x1: The left side of the p-value line to be plotted
line_x2: The right side of the p-value line to be plotted|
line_y: The height of the p-value line to be plotted
text_y: How much above the p-value line is plotted text
text: the text of p-value, you can set
***to insteadp<0.001fontsize: the fontsize of text
fontcolor: the color of text
horizontalalignment: the location of text
import seaborn as sns
data = sns.load_dataset("tips")
data.head()
total_bill tip sex smoker day time size
0 16.99 1.01 Female No Sun Dinner 2
1 10.34 1.66 Male No Sun Dinner 3
2 21.01 3.50 Male No Sun Dinner 3
3 23.68 3.31 Male No Sun Dinner 2
4 24.59 3.61 Female No Sun Dinner 4
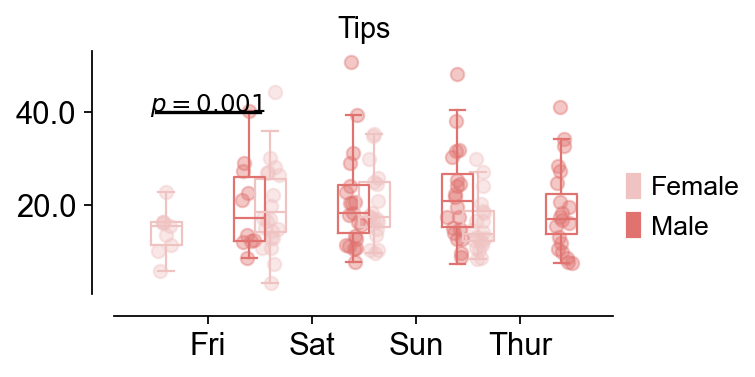
fig,ax=ov.pl.boxplot(data,hue='sex',x_value='day',y_value='total_bill',
palette=ov.pl.red_color,
figsize=(4,2),fontsize=12,title='Tips',)
ov.pl.add_palue(ax,line_x1=-0.5,line_x2=0.5,line_y=40,
text_y=0.2,
text='$p={}$'.format(round(0.001,3)),
fontsize=11,fontcolor='#000000',
horizontalalignment='center',)
📊 Boxplot Data Analysis:
Total samples: 244
X-axis variable ('day'): ['Fri', 'Sat', 'Sun', 'Thur']
Hue variable ('sex'): ['Female', 'Male']
Y-axis variable: 'total_bill' (range: 3.07 - 50.81)
⚙️ Current Function Parameters:
hue='sex', x_value='day', y_value='total_bill'
width=0.3, figsize=(4, 2), fontsize=12
hue_order=None (using alphabetical order)
📋 Using alphabetical hue order: ['Female', 'Male']
🎯 Box Positioning:
Number of hue groups: 2
Box positions: [-0.4, 0.4]
Box width: 0.3
📈 Sample sizes per group:
Female × Fri: 9 samples
Female × Sat: 28 samples
Female × Sun: 18 samples
Female × Thur: 32 samples
Male × Fri: 10 samples
Male × Sat: 59 samples
Male × Sun: 58 samples
Male × Thur: 30 samples
💡 Parameter Optimization Suggestions:
✅ Current parameters are optimal for your data!
────────────────────────────────────────────────────────────