Peak-to-gene linkage (multiome)#
A scATAC peak is only useful once you know which gene it regulates. With paired RNA + ATAC
(10x multiome) we can learn those links directly: for each peak–gene pair within a genomic
window, correlate peak accessibility against gene expression across metacells, and keep the
significant correlations. This is the ArchR / addPeak2GeneLinks idea, exposed in ov.epi as
ov.epi.tl.peak_to_gene.
This tutorial uses the public 10x PBMC 10k multiome dataset (paired scRNA + scATAC, GRCh38),
which ov.epi.datasets downloads on demand:
load the paired ATAC peak matrix + RNA matrix
build an ATAC embedding with iterative LSI
correlate peaks with genes (
ov.epi.tl.peak_to_gene)visualise the links around marker genes with
ov.epi.pl.plot_peak2gene
import warnings
warnings.filterwarnings('ignore')
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import omicverse as ov
ov.epi.pl.plot_set()
print('omicverse', ov.__version__)
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
omicverse 2.2.1rc1
1 · Load the paired multiome data#
ov.epi.datasets.pbmc10k_multiome() returns two AnnData objects sharing the same cell barcodes —
an ATAC peak matrix and an RNA count matrix, both already annotated with cell_type.
atac, rna = ov.epi.datasets.pbmc10k_multiome()
print('ATAC:', atac.shape, '| RNA:', rna.shape)
print('paired barcodes:', bool((atac.obs_names == rna.obs_names).all()))
atac.obs['cell_type'].value_counts().head()
ATAC: (9631, 107194) | RNA: (9631, 29095)
paired barcodes: True
cell_type
CD14 Mono 2554
CD4 Naive 1382
CD8 Naive 1354
CD4 TCM 1113
CD16 Mono 442
Name: count, dtype: int64
2 · Gene and exon annotation#
Both peak_to_gene (to find each gene’s TSS) and the linkage plot (to draw the gene model) need
a gene/exon annotation table. We read it from the bundled GRCh38 Gencode GFF3 with pyranges.
import pyranges as pr
gtf_path = ov.epi.data.hg38.annotation
g = pr.read_gff3(str(gtf_path)).df
def _ann(feature):
sub = g[g.Feature == feature]
return pd.DataFrame({
'gene_name': sub['gene_name'].astype(str).values,
'chrom': sub['Chromosome'].astype(str).values,
'start': sub['Start'].astype(int).values,
'end': sub['End'].astype(int).values,
'strand': sub['Strand'].astype(str).values,
})
gene_ann = _ann('gene')
exon_ann = _ann('exon')
print('genes:', len(gene_ann), '| exons:', len(exon_ann))
genes: 61852 | exons: 839796
3 · ATAC embedding#
peak_to_gene groups cells into metacells using a low-dimensional embedding; we compute the
standard ArchR-style iterative LSI on the ATAC peaks.
ov.epi.tl.iterative_lsi(atac, n_components=30, iterations=2, var_features=25000,
resolution=0.5, n_neighbors=30, seed=1)
print('ATAC embedding:', atac.obsm['X_iterative_lsi'].shape)
└─ [iterative_lsi] Initial feature set: 106,658 / 107,194
└─ [iterative_lsi] Iter 1/2 | fit on 9,631 cells x 106,658 features
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:32)
running Leiden clustering
finished: found 14 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
└─ [iterative_lsi] -> 14 clusters; selected 25,000 variable features for next round
└─ [iterative_lsi] Iter 2/2 | fit on 9,631 cells x 25,000 features
└─ [iterative_lsi] Done. Stored embedding (9,631 x 29) in adata.obsm['X_iterative_lsi']
ATAC embedding: (9631, 29)
4 · Correlate peaks with genes#
ov.epi.tl.peak_to_gene aggregates cells into metacells, then correlates each peak with every
gene whose TSS lies within max_distance (250 kb) of the peak, returning a tidy table of links
with a correlation, p-value and FDR.
links = ov.epi.tl.peak_to_gene(
atac, rna=rna, gene_annotation=gene_ann,
use_rep='X_iterative_lsi',
n_metacells=500, k_neighbors=100, max_distance=250_000, seed=1,
)
sig = links[links['fdr'] < 0.05]
print(f'{len(links):,} peak-gene pairs | {len(sig):,} significant (FDR < 0.05)')
links.head()
└─ [peak_to_gene] 107,194 peaks | 20,169 annotated genes | 9,631 cells
└─ [peak_to_gene] Building 500 metacells × 100 neighbours from X_iterative_lsi
└─ [peak_to_gene] Aggregating peak matrix
└─ [peak_to_gene] Aggregating gene matrix
└─ [peak_to_gene] Finding peak-gene pairs within ±250,000 bp
└─ [peak_to_gene] 726,292 candidate pairs
└─ [peak_to_gene] Computing correlations
└─ [peak_to_gene] 726,292 pairs retained, 441,342 significant (FDR < 0.05)
726,292 peak-gene pairs | 441,342 significant (FDR < 0.05)
| peak | gene | chrom | peak_start | peak_end | gene_start | gene_end | tss | distance | correlation | t | p_value | fdr | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | chr1:816881-817647 | LINC01409 | chr1 | 816881 | 817647 | 778746 | 810065 | 778746 | 38518 | 0.367556 | 8.819705 | 1.937570e-17 | 1.153987e-16 |
| 1 | chr1:816881-817647 | FAM87B | chr1 | 816881 | 817647 | 817370 | 819837 | 817370 | -106 | 0.214858 | 4.909412 | 1.240268e-06 | 3.701546e-06 |
| 2 | chr1:816881-817647 | LINC01128 | chr1 | 816881 | 817647 | 825137 | 859446 | 825137 | -7873 | 0.103890 | 2.331009 | 2.015113e-02 | 3.446575e-02 |
| 3 | chr1:816881-817647 | LINC00115 | chr1 | 816881 | 817647 | 824227 | 827539 | 827539 | -10275 | 0.546485 | 14.562097 | 2.831169e-40 | 3.955713e-39 |
| 4 | chr1:816881-817647 | FAM41C | chr1 | 816881 | 817647 | 868070 | 876903 | 876903 | -59639 | 0.165115 | 3.735962 | 2.086407e-04 | 4.896519e-04 |
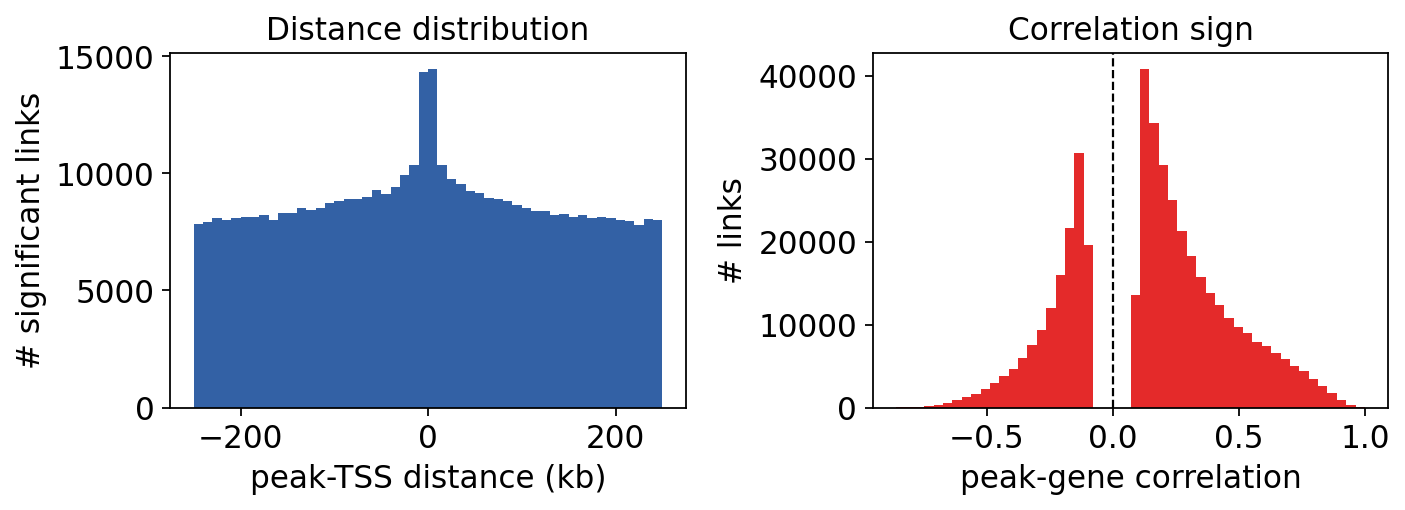
Properties of the significant links#
Two quick diagnostics: significant links are enriched at short peak–TSS distances, and positive correlations dominate (accessible enhancers tend to raise expression).
fig, axes = plt.subplots(1, 2, figsize=(9, 3.4))
axes[0].hist(sig['distance'] / 1000, bins=50, color='#3361A5')
axes[0].set_xlabel('peak-TSS distance (kb)'); axes[0].set_ylabel('# significant links')
axes[0].set_title('Distance distribution')
axes[1].hist(sig['correlation'], bins=50, color='#E42A2A')
axes[1].axvline(0, color='k', lw=1, ls='--')
axes[1].set_xlabel('peak-gene correlation'); axes[1].set_ylabel('# links')
axes[1].set_title('Correlation sign')
plt.tight_layout(); plt.show()
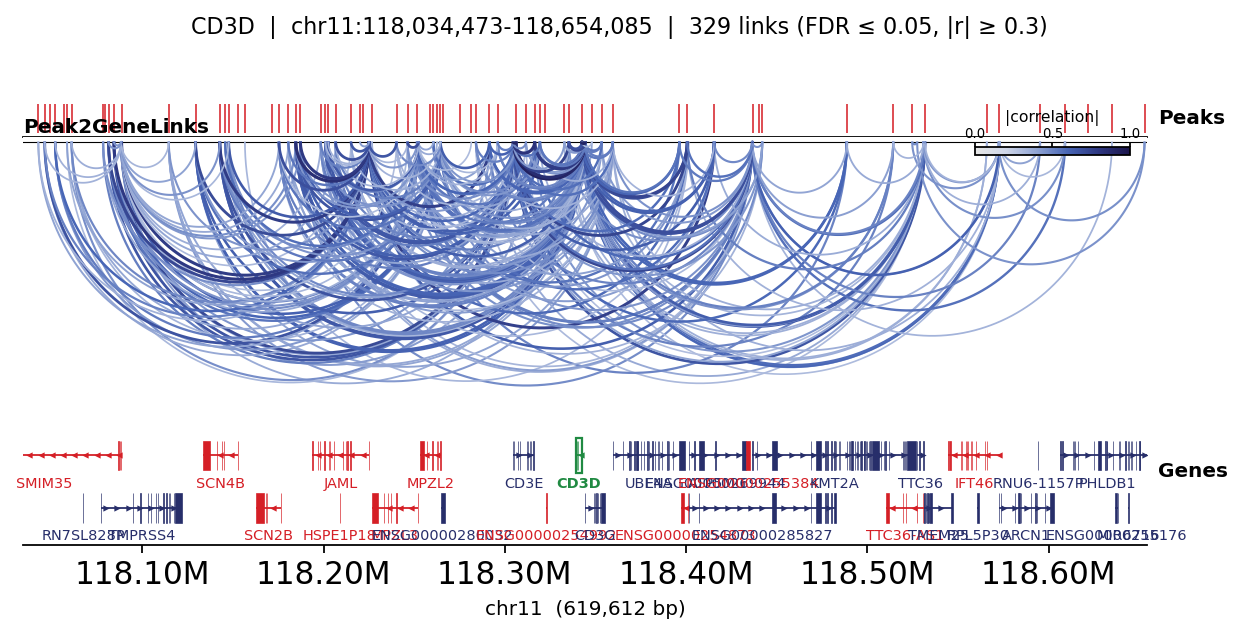
5 · Visualise links around marker genes#
ov.epi.pl.plot_peak2gene draws a genome-browser-style panel: the gene model, the peaks, and
arcs connecting each peak to the gene coloured by correlation. CD3D is a pan-T-cell gene.
fig, _ = ov.epi.pl.plot_peak2gene(
atac, gene='CD3D',
gene_annotation=gene_ann, exon_annotation=exon_ann,
fdr_thresh=0.05, min_abs_r=0.3, pad_bp=80_000, figsize=(8, 4), show=False,
)
plt.tight_layout(); plt.show()
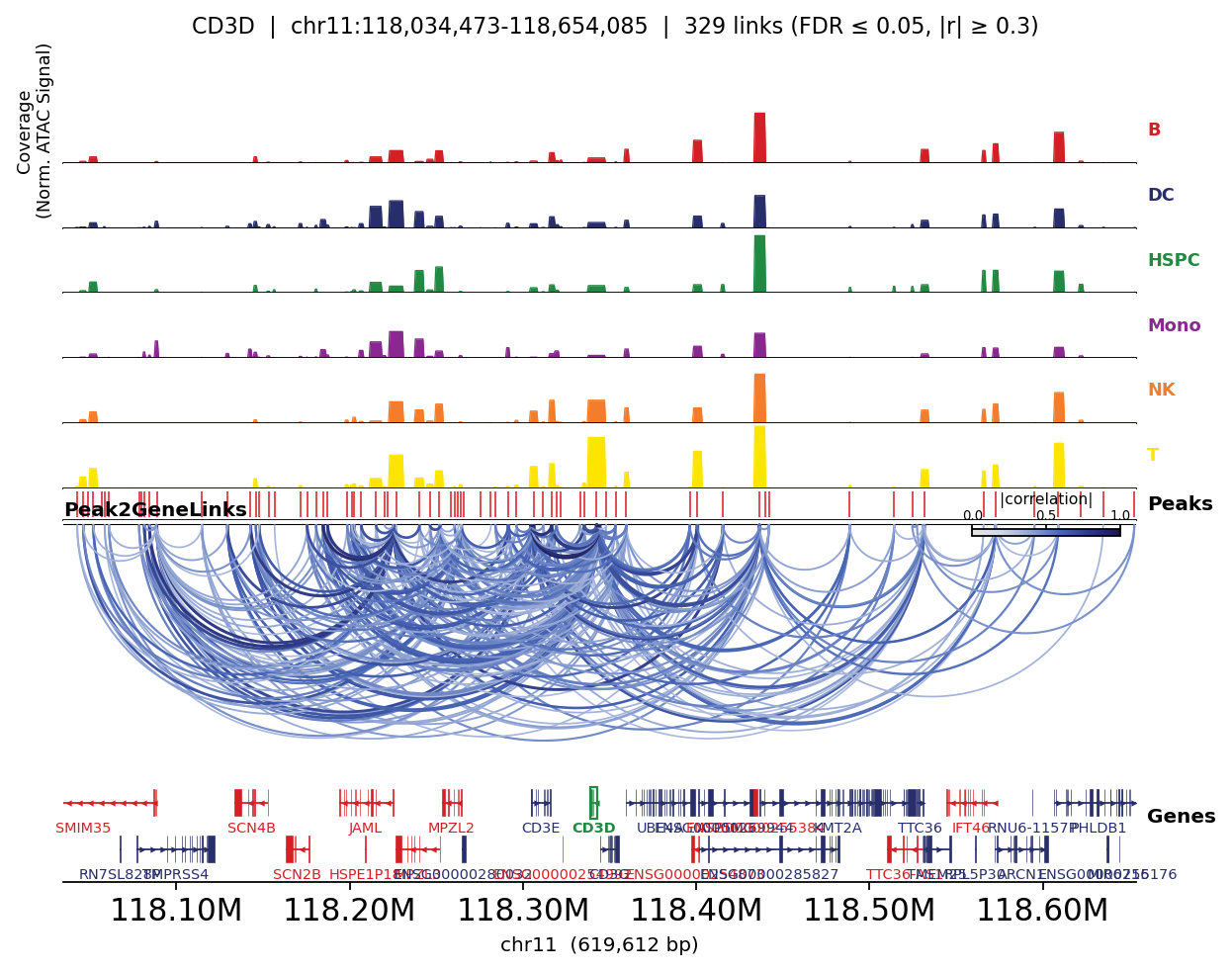
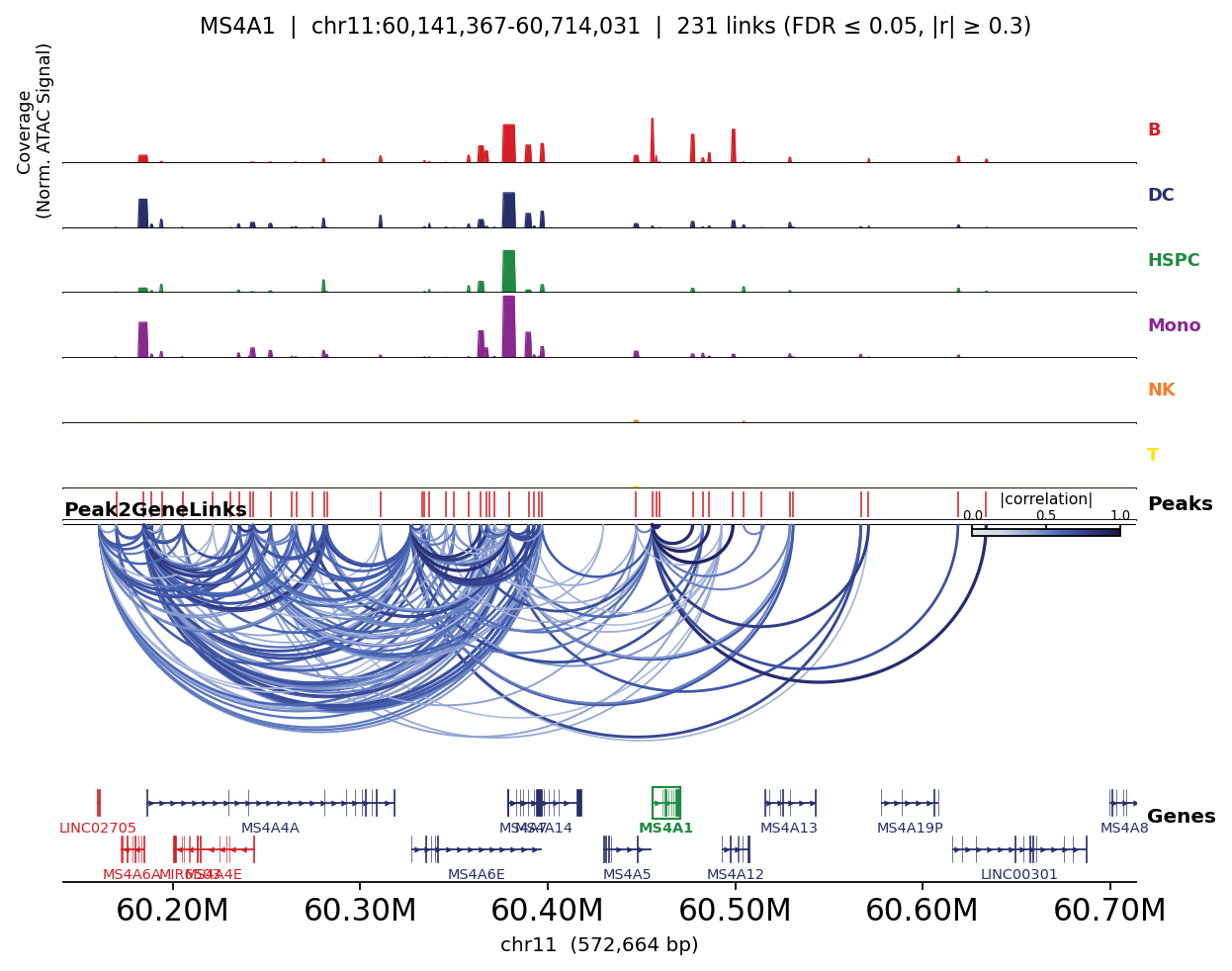
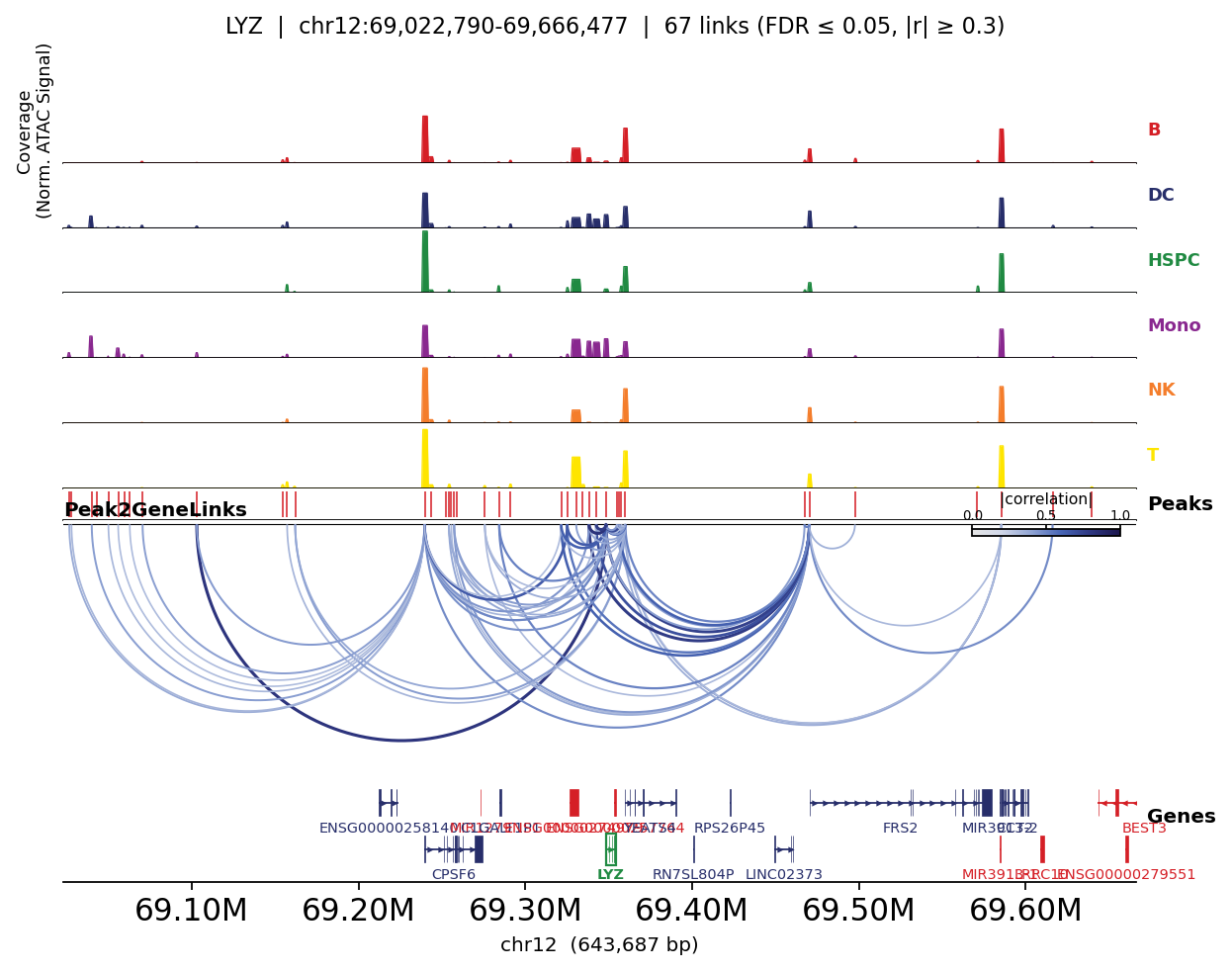
Links coloured by lineage accessibility#
Mapping the fine-grained cell_type to broad lineages lets the plot also show per-lineage peak
accessibility — so you can see, for each marker, that its linked peaks open specifically in the
matching lineage.
major = {
'CD4 Naive':'T','CD4 TCM':'T','CD4 TEM':'T','Treg':'T','CD8 Naive':'T',
'CD8 TEM_1':'T','CD8 TEM_2':'T','MAIT':'T','gdT':'T',
'Naive B':'B','Memory B':'B','Intermediate B':'B','Plasma':'B',
'NK':'NK','CD14 Mono':'Mono','CD16 Mono':'Mono','cDC':'DC','pDC':'DC','HSPC':'HSPC',
}
atac.obs['lineage'] = atac.obs['cell_type'].map(major).fillna('other').astype('category')
for gene in ['CD3D', 'MS4A1', 'LYZ']:
fig, _ = ov.epi.pl.plot_peak2gene(
atac, gene=gene, group_by='lineage',
gene_annotation=gene_ann, exon_annotation=exon_ann,
fdr_thresh=0.05, min_abs_r=0.3, pad_bp=80_000, figsize=(8, 4), show=False,
)
plt.tight_layout(); plt.show()
Summary#
stage |
function |
|---|---|
load paired RNA+ATAC |
|
ATAC embedding |
|
peak↔gene correlation |
|
linkage plot |
|
Peak-to-gene links turn an anonymous accessibility peak into a candidate regulatory element for a named gene — the foundation for enhancer-driven gene-regulatory analysis.