scRNA–scATAC integration and label transfer#
Annotating scATAC clusters by hand is hard — accessibility markers are noisier than expression.
A robust alternative is to borrow labels from a well-annotated scRNA reference: project both
modalities into a shared space (canonical correlation analysis on gene activity vs. expression),
then transfer cell-type labels by nearest neighbours. This is the ArchR
addGeneIntegrationMatrix / Seurat FindTransferAnchors idea, exposed in ov.epi as
ov.epi.tl.integrate + ov.epi.tl.transfer_labels.
We use the 10x PBMC 10k multiome dataset. Because it is paired (RNA + ATAC measured in the same cells), it gives us a rare luxury: ground-truth cell types for the ATAC cells, so we can measure how accurate the transferred labels are.
build an ATAC gene-activity matrix from fragments
CCA-integrate it with the RNA reference (
ov.epi.tl.integrate)transfer RNA cell-type labels onto the ATAC cells (
ov.epi.tl.transfer_labels)score the transfer against the held-out true labels and visualise the joint embedding
Runtime: building the gene-activity matrix imports a ~2 GB fragments file and tiles the genome for ~13k cells — a few minutes of CPU. Everything is cached for re-runs.
import warnings
warnings.filterwarnings('ignore')
import os
import numpy as np
import pandas as pd
import anndata as ad
import scanpy as sc
import matplotlib.pyplot as plt
import omicverse as ov
ov.epi.pl.plot_set()
print('omicverse', ov.__version__)
└─ 🔬 Starting plot initialization...
├─ Apply Scanpy/matplotlib settings
├─ Custom font setup
├─ Suppress warnings
├─
___________ .__
\_ _____/_____ |__| ____ ____ ____
| __)_\____ \| |/ _ \ / \_/ __ \
| \ |_> > ( <_> ) | \ ___/
/_______ / __/|__|\____/|___| /\___ >
\/|__| \/ \/
├─ 🔖 Version: 0.0.1rc1 📚 Tutorials: https://epione.readthedocs.io/
└─ ✅ plot_set complete.
omicverse 2.2.1rc1
1 · Reference RNA + ATAC gene activity#
The RNA reference is the annotated multiome RNA matrix. For the ATAC query we need a matrix in
gene space: we import the ATAC fragments, build a tile matrix, and aggregate it into per-gene
activity scores (ov.epi.tl.add_gene_score_matrix).
genome = ov.epi.data.hg38
genes = ov.epi.io.get_gene_annotation(genome)
atac_peaks, rna = ov.epi.datasets.pbmc10k_multiome() # peak matrix (for true labels) + RNA
frag = ov.epi.datasets.pbmc10k_atac_fragments()
work = os.path.join(os.getcwd(), 'data_epi')
os.makedirs(work, exist_ok=True)
h5 = os.path.join(work, 'atac10k_geneact.h5ad')
if os.path.exists(h5):
os.remove(h5)
atac = ov.epi.pp.import_fragments(str(frag), chrom_sizes=genome, file=h5, min_num_fragments=500)
ov.epi.pp.add_tile_matrix(atac, bin_size=500)
ov.epi.tl.add_gene_score_matrix(atac, genes)
print('ATAC gene-activity:', np.asarray(atac.obsm['gene_score']).shape)
└─ scanning /tmp/snapatac2/pbmc_10k_atac.tsv.gz
└─ imported 13,336 cells (182,364,051 unique fragments)
└─ building tile matrix: 13,336 cells × 6,176,584 tiles (500 bp bins, strategy=paired-insertion)
└─ tile matrix nnz=221,104,147
└─ [gene_score] genes: 19,923 on 23 chroms (after excluding ['chrY', 'chrM'])
└─ [gene_score] chrom-by-chrom ArchR path (fragments → uniq_tiles → W @ matGS)
└─ [gene_score] chr=chr1 genes=2,061
└─ [gene_score] chr=chr10 genes=730
└─ [gene_score] chr=chr11 genes=1,318
└─ [gene_score] chr=chr12 genes=1,037
└─ [gene_score] chr=chr13 genes=322
└─ [gene_score] chr=chr14 genes=615
└─ [gene_score] chr=chr15 genes=600
└─ [gene_score] chr=chr16 genes=856
└─ [gene_score] chr=chr17 genes=1,185
└─ [gene_score] chr=chr18 genes=266
└─ [gene_score] chr=chr19 genes=1,474
└─ [gene_score] chr=chr2 genes=1,247
└─ [gene_score] chr=chr20 genes=544
└─ [gene_score] chr=chr21 genes=219
└─ [gene_score] chr=chr22 genes=446
└─ [gene_score] chr=chr3 genes=1,077
└─ [gene_score] chr=chr4 genes=753
└─ [gene_score] chr=chr5 genes=881
└─ [gene_score] chr=chr6 genes=1,049
└─ [gene_score] chr=chr7 genes=929
└─ [gene_score] chr=chr8 genes=695
└─ [gene_score] chr=chr9 genes=774
└─ [gene_score] chr=chrX genes=845
└─ [gene_score] done. obsm['gene_score'] = (13336, 19923) float32 (uns[gene_score_gene_names]: 19,923 genes)
ATAC gene-activity: (13336, 19923)
Wrap gene activity as a gene-space AnnData#
We move the gene-score matrix into X of a new AnnData whose var_names are gene symbols, and
attach each ATAC cell’s true cell type (from the paired peak-matrix object) so we can grade
the transfer later.
gs = np.asarray(atac.obsm['gene_score'])
gnames = [str(x) for x in np.asarray(atac.uns['gene_score_gene_names'])]
atac_ga = ad.AnnData(X=gs)
atac_ga.var_names = gnames
atac_ga.obs_names = list(atac.obs_names)
common = atac_ga.obs_names.intersection(atac_peaks.obs_names)
atac_ga = atac_ga[common].copy()
atac_ga.obs['cell_type_true'] = atac_peaks.obs.loc[common, 'cell_type'].values
print('ATAC cells with ground-truth labels:', atac_ga.n_obs)
ATAC cells with ground-truth labels: 9631
3 · Transfer labels and score the result#
ov.epi.tl.transfer_labels does weighted kNN voting in the shared space, writing the predicted
label to obs['celltype'] and a confidence to obs['transfer_score'].
ov.epi.tl.transfer_labels(atac_ga, rna, reference_label='cell_type', k=30)
pred = atac_ga.obs['celltype'].astype(str).values
true = atac_ga.obs['cell_type_true'].astype(str).values
acc = float((pred == true).mean())
print(f'label-transfer accuracy vs ground truth: {acc:.1%}')
atac_ga.obs[['celltype', 'cell_type_true', 'transfer_score']].head()
label-transfer accuracy vs ground truth: 83.3%
| celltype | cell_type_true | transfer_score | |
|---|---|---|---|
| AAACAGCCAATCCCTT-1 | CD4 TEM | CD4 TCM | 0.358502 |
| AAACAGCCAATGCGCT-1 | CD4 Naive | CD4 Naive | 0.783197 |
| AAACAGCCACCAACCG-1 | CD8 Naive | CD8 Naive | 0.303284 |
| AAACAGCCAGGATAAC-1 | CD4 Naive | CD4 Naive | 0.868715 |
| AAACAGCCAGTTTACG-1 | CD4 TCM | CD4 TCM | 0.715788 |
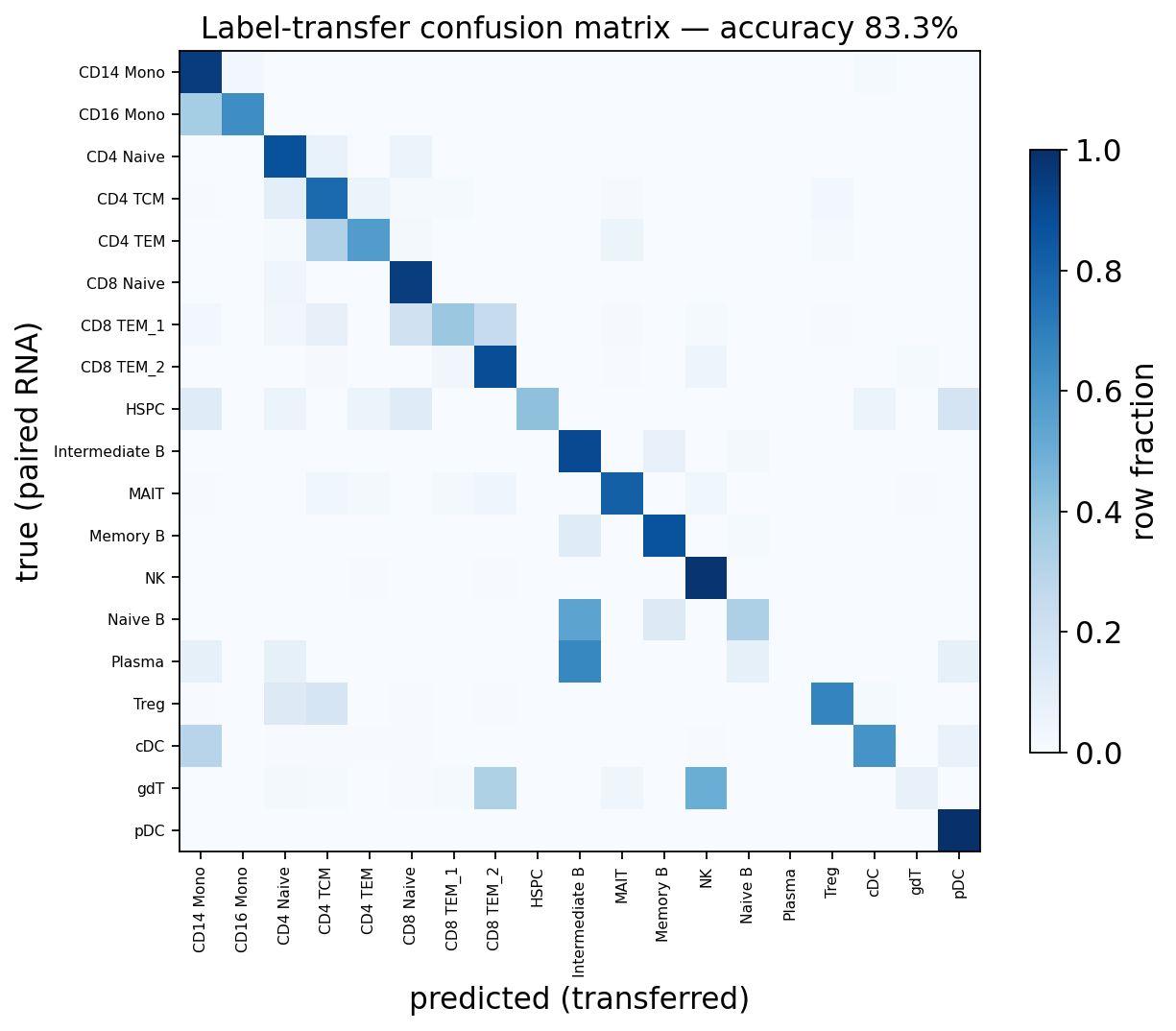
Confusion matrix#
A row-normalised confusion matrix of predicted vs. true cell type — a strong diagonal means the transfer recovers the right identities.
cts = sorted(set(true) | set(pred))
cm = pd.crosstab(pd.Series(true, name='true'), pd.Series(pred, name='pred'))
cm = cm.reindex(index=cts, columns=cts, fill_value=0)
cmn = cm.div(cm.sum(1).replace(0, 1), axis=0)
fig, ax = plt.subplots(figsize=(8, 7))
im = ax.imshow(cmn.values, cmap='Blues', vmin=0, vmax=1)
ax.set_xticks(range(len(cts))); ax.set_xticklabels(cts, rotation=90, fontsize=7)
ax.set_yticks(range(len(cts))); ax.set_yticklabels(cts, fontsize=7)
ax.set_xlabel('predicted (transferred)'); ax.set_ylabel('true (paired RNA)')
ax.set_title(f'Label-transfer confusion matrix — accuracy {acc:.1%}')
plt.colorbar(im, ax=ax, label='row fraction', shrink=0.7)
plt.tight_layout(); plt.show()
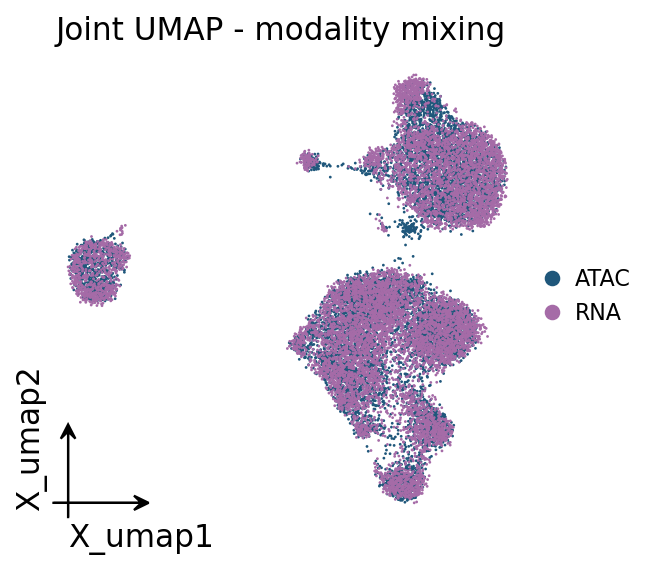
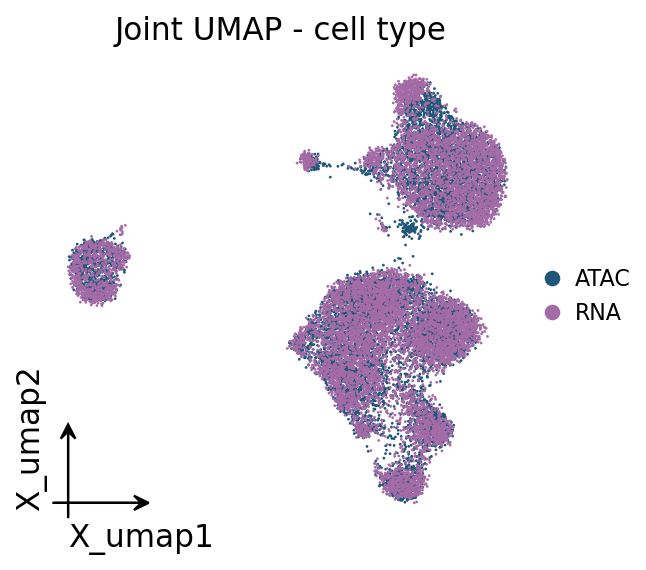
4 · Joint embedding#
ov.epi.tl.joint_embedding concatenates both modalities on the shared CCA space and runs a
UMAP, so we can confirm the two assays overlap (good mixing) and that transferred labels are
spatially coherent.
joint = ov.epi.tl.joint_embedding(
atac_ga, rna, use_rep='X_cca', labels=('ATAC', 'RNA'),
n_neighbors=30, metric='cosine', random_state=0,
)
# Joint embedding with omicverse's plotter: modality mixing + cell type.
ov.pl.embedding(joint, basis='X_umap', color='modality', frameon='small',
title='Joint UMAP - modality mixing', show=False)
plt.show()
lab_key = 'celltype_joint' if 'celltype_joint' in joint.obs else (
'celltype' if 'celltype' in joint.obs else 'modality')
ov.pl.embedding(joint, basis='X_umap', color=lab_key, frameon='small',
title='Joint UMAP - cell type', show=False)
plt.show()
computing neighbors
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:33)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm)
'umap', UMAP parameters (adata.uns) (0:00:15)
Summary#
stage |
function |
|---|---|
ATAC gene activity |
|
CCA integration |
|
label transfer |
|
joint embedding |
|
Transferring labels from a paired RNA reference recovered the ATAC cell identities with ~83 % accuracy here — and on unpaired data (separate scRNA and scATAC experiments) the exact same calls give you an annotated scATAC dataset without any manual marker curation.