Batch correction with Harmony#
Harmony is the workhorse PCA-embedding integration method (Korsunsky et al., Nat Methods 2019). It runs entirely on the low-dimensional embedding, never touches the expression matrix, is the fastest of the lot, and is the only backend that works out-of-core.
This is one of the omicverse batch-correction zoo tutorials. See batch/index for the overview / decision tree, or ../t_single_batch for the side-by-side comparison of every backend on a real benchmark.
Load a 2-batch demo from pbmc3k#
We use the canonical 10x pbmc3k dataset and synthesise a 2-batch label by random assignment, then plant a gene-shift on batch_B so the uncorrected UMAP shows a visible batch effect. This keeps the notebook self-contained and fast (~2 min end-to-end) — for a real multi-donor benchmark with [scib-metrics] scoring, see ../t_single_batch.
import omicverse as ov
ov.style()
import anndata as ad
import numpy as np
import pandas as pd
# NeurIPS 2021 multimodal hematopoiesis dataset — 3 real donor batches
# (s1d3, s2d1, s3d7), pre-annotated `cell_type` and raw `layers['counts']`.
# Same datasets used by the overview notebook ../t_single_batch.ipynb.
adata1 = ov.datasets.get_adata(
'https://figshare.com/ndownloader/files/41932005',
filename='neurips2021_s1d3.h5ad',
)
adata2 = ov.datasets.get_adata(
'https://figshare.com/ndownloader/files/41932008',
filename='neurips2021_s2d1.h5ad',
)
adata3 = ov.datasets.get_adata(
'https://figshare.com/ndownloader/files/41932011',
filename='neurips2021_s3d7.h5ad',
)
adata = ad.concat([adata1, adata2, adata3], merge='same')
adata.var_names_make_unique()
adata.obs_names_make_unique()
adata.obs['batch'] = adata.obs['batch'].astype('category')
# Subsample to a quick-running ~6 000 cells × 3 batches so the CPU
# backends (harmony / combat / scanorama / cca) finish in well under a
# minute. Drop this line for a full-resolution run.
_rng = np.random.default_rng(0)
_sel = _rng.choice(adata.n_obs, 6000, replace=False)
adata = adata[_sel].copy()
adata
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
✅ NVIDIA CUDA GPUs detected: 1
• [CUDA 0] NVIDIA H100 80GB HBM3
Memory: 79.1 GB | Compute: 9.0
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
⚠️ File ./data/neurips2021_s1d3.h5ad already exists
Loading data from ./data/neurips2021_s1d3.h5ad
✅ Successfully loaded: 5935 cells × 13953 genes
⚠️ File ./data/neurips2021_s2d1.h5ad already exists
Loading data from ./data/neurips2021_s2d1.h5ad
✅ Successfully loaded: 10258 cells × 13953 genes
⚠️ File ./data/neurips2021_s3d7.h5ad already exists
Loading data from ./data/neurips2021_s3d7.h5ad
✅ Successfully loaded: 11230 cells × 13953 genes
AnnData object with n_obs × n_vars = 6000 × 13953
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train'
var: 'feature_types', 'gene_id'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap'
layers: 'counts'
Preprocess + PCA + cluster#
Same QC → HVG-pearson → log-norm → PCA pipeline shared across every backend in the zoo. A quick Leiden cluster gives a synthetic celltype label that scANVI / scPoli can use as a prototype anchor.
# Standard omicverse preprocess (QC → HVG-via-pearson → log-norm → PCA).
# QC thresholds are loose because the NeurIPS data is already filtered.
adata = ov.pp.qc(adata, tresh={'mito_perc': 0.2, 'nUMIs': 200,
'detected_genes': 100})
ov.utils.store_layers(adata, layers='counts')
adata = ov.pp.preprocess(adata, mode='shiftlog|pearson', n_HVGs=2000,
batch_key=None)
adata.raw = adata
adata = adata[:, adata.var.highly_variable_features].copy()
ov.pp.scale(adata)
ov.pp.pca(adata, layer='scaled', n_pcs=30)
# Neighbours graph for the pre-correction UMAP.
ov.pp.neighbors(adata, use_rep='scaled|original|X_pca', n_neighbors=15)
# The NeurIPS adata already carries a real `cell_type` annotation —
# rename it to `celltype` for the wrapper's expected schema. No Leiden
# needed; the labels are pre-annotated by the dataset authors.
adata.obs['celltype'] = adata.obs['cell_type'].astype('category')
adata
🖥️ Using CPU mode for QC...
Auto-detected mitochondrial prefix: 'MT-'
📊 Step 1: Calculating QC Metrics
✓ Gene Family Detection:
┌──────────────────────────────┬────────────────────┬────────────────────┐
│ Gene Family │ Genes Found │ Detection Method │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Mitochondrial │ 13 │ Auto (MT-) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Ribosomal │ 94 │ Auto (RPS/RPL) │
├──────────────────────────────┼────────────────────┼────────────────────┤
│ Hemoglobin │ 11 │ Auto (regex) │
└──────────────────────────────┴────────────────────┴────────────────────┘
✓ QC Metrics Summary:
┌─────────────────────────┬────────────────────┬─────────────────────────┐
│ Metric │ Mean │ Range (Min - Max) │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ nUMIs │ 20693 │ 1999 - 627052 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Detected Genes │ 1346 │ 104 - 5543 │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Mitochondrial % │ 6.6% │ 0.0% - 20.0% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Ribosomal % │ 24.5% │ 0.1% - 63.4% │
├─────────────────────────┼────────────────────┼─────────────────────────┤
│ Hemoglobin % │ 10.1% │ 0.0% - 96.1% │
└─────────────────────────┴────────────────────┴─────────────────────────┘
📈 Original cell count: 6,000
🔧 Step 2: Quality Filtering (SEURAT)
Thresholds: mito≤0.2, nUMIs≥200, genes≥100
📊 Seurat Filter Results:
• nUMIs filter (≥200): 0 cells failed (0.0%)
• Genes filter (≥100): 0 cells failed (0.0%)
• Mitochondrial filter (≤0.2): 1 cells failed (0.0%)
✓ Filters applied successfully
✓ Combined QC filters: 1 cells removed (0.0%)
🎯 Step 3: Final Filtering
Parameters: min_genes=200, min_cells=3
Ratios: max_genes_ratio=1, max_cells_ratio=1
✓ Final filtering: 44 cells, 0 genes removed
🔍 Step 4: Doublet Detection
💡 Running pyscdblfinder (Python port of R scDblFinder)
🔍 Running scdblfinder detection...
[ScDblFinder] wrote scDblFinder_score + scDblFinder_class — threshold=0.040
✓ scDblFinder completed: 169 doublets removed (2.8%)
╭─ SUMMARY: qc ──────────────────────────────────────────────────────╮
│ Duration: 27.9406s │
│ Shape: 6,000 x 13,953 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBS │ ✚ cell_complexity (float) │
│ │ ✚ detected_genes (int) │
│ │ ✚ hb_perc (float) │
│ │ ✚ mito_perc (float) │
│ │ ✚ nUMIs (float) │
│ │ ✚ n_counts (float) │
│ │ ✚ n_genes (int) │
│ │ ✚ n_genes_by_counts (int) │
│ │ ✚ passing_mt (bool) │
│ │ ✚ passing_nUMIs (bool) │
│ │ ✚ passing_ngenes (bool) │
│ │ ✚ pct_counts_hb (float) │
│ │ ✚ pct_counts_mt (float) │
│ │ ✚ pct_counts_ribo (float) │
│ │ ✚ ribo_perc (float) │
│ │ ✚ total_counts (float) │
│ │
│ ● VAR │ ✚ hb (bool) │
│ │ ✚ mt (bool) │
│ │ ✚ ribo (bool) │
│ │
╰────────────────────────────────────────────────────────────────────╯
......The X of adata have been stored in counts
🔍 [2026-05-29 04:54:20] Running preprocessing in 'cpu' mode...
Begin robust gene identification
After filtration, 13953/13953 genes are kept.
Among 13953 genes, 13953 genes are robust.
✅ Robust gene identification completed successfully.
Begin size normalization: shiftlog and HVGs selection pearson
🔍 Count Normalization:
Target sum: 500000.0
Exclude highly expressed: True
Max fraction threshold: 0.2
⚠️ Excluding 6 highly-expressed genes from normalization computation
Excluded genes: ['IGKC', 'HBB', 'MALAT1', 'HBA2', 'IGLC2', 'IGLC3']
✅ Count Normalization Completed Successfully!
✓ Processed: 5,786 cells × 13,953 genes
✓ Runtime: 0.28s
🔍 Highly Variable Genes Selection (Experimental):
Method: pearson_residuals
Target genes: 2,000
Theta (overdispersion): 100
✅ Experimental HVG Selection Completed Successfully!
✓ Selected: 2,000 highly variable genes out of 13,953 total (14.3%)
✓ Results added to AnnData object:
• 'highly_variable': Boolean vector (adata.var)
• 'highly_variable_rank': Float vector (adata.var)
• 'highly_variable_nbatches': Int vector (adata.var)
• 'highly_variable_intersection': Boolean vector (adata.var)
• 'means': Float vector (adata.var)
• 'variances': Float vector (adata.var)
• 'residual_variances': Float vector (adata.var)
Time to analyze data in cpu: 2.00 seconds.
✅ Preprocessing completed successfully.
Added:
'highly_variable_features', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
'counts', raw counts layer (adata.layers)
End of size normalization: shiftlog and HVGs selection pearson
╭─ SUMMARY: preprocess ──────────────────────────────────────────────╮
│ Duration: 2.1728s │
│ Shape: 5,786 x 13,953 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● VAR │ ✚ highly_variable (bool) │
│ │ ✚ highly_variable_features (bool) │
│ │ ✚ highly_variable_rank (float) │
│ │ ✚ means (float) │
│ │ ✚ n_cells (int) │
│ │ ✚ percent_cells (float) │
│ │ ✚ residual_variances (float) │
│ │ ✚ robust (bool) │
│ │ ✚ variances (float) │
│ │
│ ● UNS │ ✚ history_log │
│ │ ✚ hvg │
│ │ ✚ log1p │
│ │
╰────────────────────────────────────────────────────────────────────╯
╭─ SUMMARY: scale ───────────────────────────────────────────────────╮
│ Duration: 0.143s │
│ Shape: 5,786 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● LAYERS │ ✚ scaled (array, 5786x2000) │
│ │
╰────────────────────────────────────────────────────────────────────╯
computing PCA🔍
with n_comps=30
🖥️ Using sklearn PCA for CPU computation
🖥️ sklearn PCA backend: CPU computation
📊 PCA input data type: ArrayView, shape: (5786, 2000), dtype: float64
🔧 PCA solver used: covariance_eigh
finished✅ (1.16s)
╭─ SUMMARY: pca ─────────────────────────────────────────────────────╮
│ Duration: 1.1684s │
│ Shape: 5,786 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ pca │
│ │ └─ params: {'zero_center': True, 'use_highly_variable': Tr...│
│ │ ✚ scaled|original|cum_sum_eigenvalues │
│ │ ✚ scaled|original|pca_var_ratios │
│ │
│ ● OBSM │ ✚ X_pca (array, 5786x30) │
│ │ ✚ scaled|original|X_pca (array, 5786x30) │
│ │
╰────────────────────────────────────────────────────────────────────╯
🖥️ Using Scanpy CPU to calculate neighbors...
🔍 K-Nearest Neighbors Graph Construction:
Mode: cpu
Neighbors: 15
Method: umap
Metric: euclidean
Representation: scaled|original|X_pca
🔍 Computing neighbor distances...
🔍 Computing connectivity matrix...
💡 Using UMAP-style connectivity
✓ Graph is fully connected
✅ KNN Graph Construction Completed Successfully!
✓ Processed: 5,786 cells with 15 neighbors each
✓ Results added to AnnData object:
• 'neighbors': Neighbors metadata (adata.uns)
• 'distances': Distance matrix (adata.obsp)
• 'connectivities': Connectivity matrix (adata.obsp)
╭─ SUMMARY: neighbors ───────────────────────────────────────────────╮
│ Duration: 8.4605s │
│ Shape: 5,786 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ neighbors │
│ │ └─ params: {'n_neighbors': 15, 'method': 'umap', 'random_s...│
│ │
│ ● OBSP │ ✚ connectivities (sparse matrix, 5786x5786) │
│ │ ✚ distances (sparse matrix, 5786x5786) │
│ │
╰────────────────────────────────────────────────────────────────────╯
AnnData object with n_obs × n_vars = 5786 × 2000
obs: 'GEX_n_genes_by_counts', 'GEX_pct_counts_mt', 'GEX_size_factors', 'GEX_phase', 'ADT_n_antibodies_by_counts', 'ADT_total_counts', 'ADT_iso_count', 'cell_type', 'batch', 'ADT_pseudotime_order', 'GEX_pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', 'is_train', 'nUMIs', 'mito_perc', 'ribo_perc', 'hb_perc', 'detected_genes', 'cell_complexity', 'n_counts', 'total_counts', 'n_genes', 'n_genes_by_counts', 'pct_counts_mt', 'pct_counts_ribo', 'pct_counts_hb', 'passing_mt', 'passing_nUMIs', 'passing_ngenes', 'predicted_doublet', 'doublet_score', 'scdblfinder_doublet', 'scdblfinder_score', 'celltype'
var: 'feature_types', 'gene_id', 'mt', 'ribo', 'hb', 'n_cells', 'percent_cells', 'robust', 'highly_variable_features', 'means', 'variances', 'residual_variances', 'highly_variable_rank', 'highly_variable'
uns: 'status', 'status_args', 'REFERENCE_MANU', '_ov_provenance', 'layers_counts', 'history_log', 'log1p', 'hvg', 'pca', 'scaled|original|pca_var_ratios', 'scaled|original|cum_sum_eigenvalues', 'neighbors'
obsm: 'ADT_X_pca', 'ADT_X_umap', 'ADT_isotype_controls', 'GEX_X_pca', 'GEX_X_umap', 'X_pca', 'scaled|original|X_pca'
varm: 'PCs', 'scaled|original|pca_loadings'
layers: 'counts', 'scaled'
obsp: 'distances', 'connectivities'
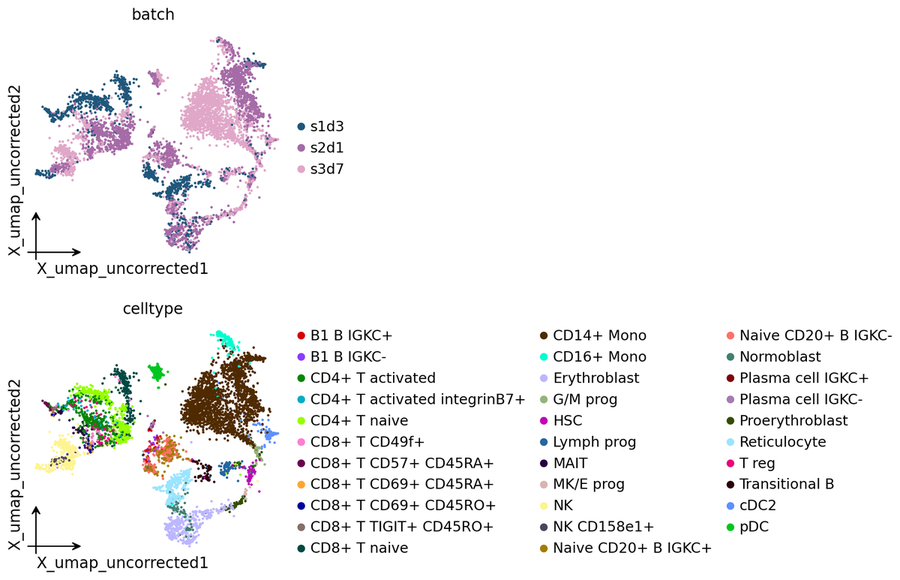
Uncorrected baseline#
The planted batch effect is visible in the uncorrected UMAP:
# Pre-correction UMAP shows the planted batch effect.
ov.pp.umap(adata, min_dist=0.3)
adata.obsm['X_umap_uncorrected'] = adata.obsm['X_umap'].copy()
ov.pl.embedding(adata, basis='X_umap_uncorrected',
color=['batch', 'celltype'],
frameon='small', wspace=0.5)
🔍 [2026-05-29 04:54:32] Running UMAP in 'cpu' mode...
🖥️ Using Scanpy CPU UMAP...
🔍 UMAP Dimensionality Reduction:
Mode: cpu
Method: umap
Components: 2
Min distance: 0.3
{'n_neighbors': 15, 'method': 'umap', 'random_state': 0, 'metric': 'euclidean', 'use_rep': 'scaled|original|X_pca'}
🔍 Computing UMAP parameters...
🔍 Computing UMAP embedding (classic method)...
✅ UMAP Dimensionality Reduction Completed Successfully!
✓ Embedding shape: 5,786 cells × 2 dimensions
✓ Results added to AnnData object:
• 'X_umap': UMAP coordinates (adata.obsm)
• 'umap': UMAP parameters (adata.uns)
✅ UMAP completed successfully.
╭─ SUMMARY: umap ────────────────────────────────────────────────────╮
│ Duration: 0.9425s │
│ Shape: 5,786 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● UNS │ ✚ umap │
│ │ └─ params: {'a': np.float64(0.9921756195894755), 'b': np.f...│
│ │
│ ● OBSM │ ✚ X_umap (array, 5786x2) │
│ │
╰────────────────────────────────────────────────────────────────────╯
Run ov.single.batch_correction(methods='harmony')#
For the scvi-tools family backends, the wrapper auto-routes **kwargs between the model’s __init__ (architecture) and .train() (optimisation) destinations. See the Key parameters section below.
ov.single.batch_correction(
adata,
batch_key='batch',
methods='harmony',
n_pcs=30,
)
...Begin using harmony to correct batch effect
🚀 Using PyTorch CUDA acceleration for Harmony
Data: 30 PCs × 5786 cells
Batch variables: ['batch']
Max iterations: 10
Convergence threshold: 0.0001
Initializing centroids (K=100) ...
done
🔍 [2026-05-29 04:54:39] Running Harmony integration...
✅ Harmony converged after 5 iterations
╭─ SUMMARY: batch_correction ────────────────────────────────────────╮
│ Duration: 1.7246s │
│ Shape: 5,786 x 2,000 (Unchanged) │
│ │
│ CHANGES DETECTED │
│ ──────────────── │
│ ● OBSM │ ✚ X_harmony (array, 5786x30) │
│ │ ✚ X_pca_harmony (array, 5786x30) │
│ │
╰────────────────────────────────────────────────────────────────────╯
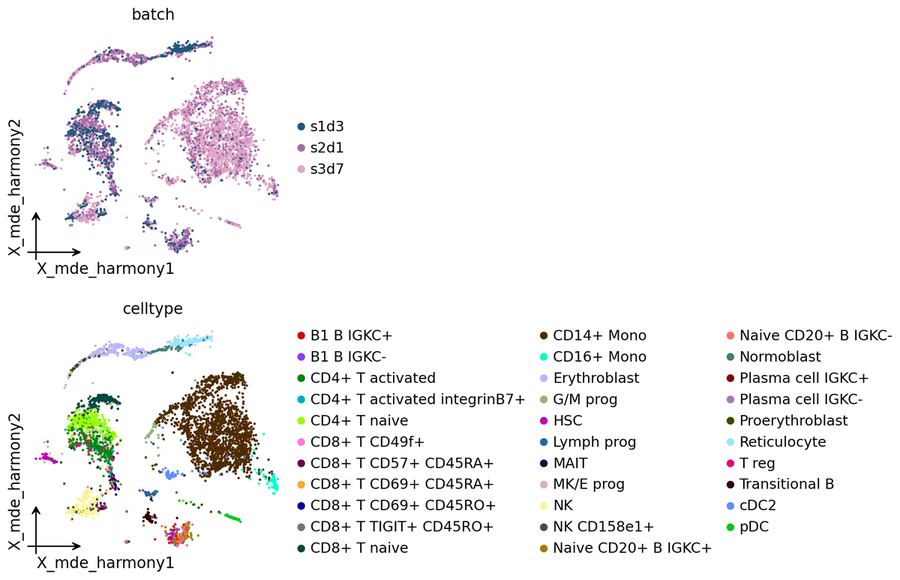
Corrected embedding#
Every backend writes its corrected representation to a stable obsm key — for this one it is adata.obsm['X_pca_harmony']. We project via ov.utils.mde for a lightweight UMAP-style display.
adata.obsm['X_mde_harmony'] = ov.utils.mde(adata.obsm['X_pca_harmony'])
ov.pl.embedding(
adata,
basis='X_mde_harmony',
color=['batch', 'celltype'],
frameon='small',
wspace=0.5,
)
Key parameters#
n_pcs— number of PCs Harmony iterates over (default 50).theta(advanced, via**kwargs) — diversity-clustering penalty.use_rep— name of the input PCA embedding to correct.