Lipidomics — the lipidr workflow in omicverse#
Lipidomics has enough structure to deserve a dedicated analysis path. Every
lipid follows the LIPID MAPS shorthand (PE 34:1, TAG 54:3,
Cer d18:1/24:0), targeted assays report several transitions per lipid,
and the biology is organised by class, chain length and
unsaturation — none of which a generic metabolomics pipeline exploits.
omicverse integrates the Bioconductor lipidr
workflow through the pure-Python pylipidr
backend. Every step is a registered ov.metabol function and every object is
a plain AnnData, so lipidomics drops straight into the rest of omicverse.
Pipeline. read_skyline → add_sample_annotation → summarize_transitions
→ annotate_lipids → normalize_pqn → de_lipids → lsea / lipid_mva.
Install the backend with
pip install omicverse[lipidomics].
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import omicverse as ov
ov.plot_set()
🔬 Starting plot initialization...
🧬 Detecting GPU devices…
🚫 No GPU devices found (CUDA/MPS/ROCm/XPU)
____ _ _ __
/ __ \____ ___ (_)___| | / /__ _____________
/ / / / __ `__ \/ / ___/ | / / _ \/ ___/ ___/ _ \
/ /_/ / / / / / / / /__ | |/ / __/ / (__ ) __/
\____/_/ /_/ /_/_/\___/ |___/\___/_/ /____/\___/
🔖 Version: 2.2.1rc1 📚 Tutorials: https://omicverse.readthedocs.io/
✅ plot_set complete.
1 — Download a raw Skyline dataset#
We use lipidr’s own targeted-lipidomics example — a mouse diet study
(Salek/Mohamed): liver lipids measured by LC-MS/MS, exported from Skyline.
Two MIT-licensed files live on GitHub, so nothing is bundled locally:
A1_data.csv— the long Skyline transition table (one row per transition)clin.csv— sample metadata:group,Diet(Normal / HighFat),BileAcid
ov.datasets.download_data caches each file under ./metabol_data.
base = 'https://raw.githubusercontent.com/ahmohamed/lipidr/master/inst/extdata'
skyline_path = ov.datasets.download_data(
url=f'{base}/A1_data.csv', file_path='A1_data.csv', dir='./metabol_data',
)
clin_path = ov.datasets.download_data(
url=f'{base}/clin.csv', file_path='clin.csv', dir='./metabol_data',
)
🔍 Downloading data to ./metabol_data/A1_data.csv
⚠️ File ./metabol_data/A1_data.csv already exists
🔍 Downloading data to ./metabol_data/clin.csv
⚠️ File ./metabol_data/clin.csv already exists
2 — Read the Skyline export#
ov.metabol.read_skyline pivots the long transition table into an
AnnData (samples × transitions). ov.metabol.add_sample_annotation
joins the clinical table onto adata.obs by sample id.
adata = ov.metabol.read_skyline(skyline_path)
adata = ov.metabol.add_sample_annotation(adata, clin_path)
print(adata)
print('groups:', adata.obs['group'].value_counts().to_dict())
AnnData object with n_obs × n_vars = 58 × 102
obs: 'group', 'Diet', 'BileAcid'
var: 'Molecule', 'TransitionId', 'Class_skyline', 'Class', 'Category', 'total_cl', 'total_cs', 'chains', 'not_parsed', 'istd'
uns: 'lipidr_default_measure', 'lipidr_state'
groups: {'QC': 12, 'NormalDiet_water': 11, 'NormalDiet_DCA': 11, 'HighFat_water': 11, 'HighFat_DCA': 11, 'blank': 2}
3 — Summarize transitions#
A targeted assay records several transitions per lipid (different
product ions). summarize_transitions collapses them to one value per
lipid — method='max' keeps the most intense transition, the lipidr
default and the most robust choice for quantification.
adata = ov.metabol.summarize_transitions(adata, method='max')
print('after summarization:', adata.shape, '(samples × lipids)')
after summarization: (58, 101) (samples × lipids)
4 — Annotate lipids with Goslin#
ov.metabol.annotate_lipids parses every lipid name with the Goslin
reference engine (pygoslin) and writes lipid_class, lipid_category,
total_carbons, total_db and lipid_backbone into adata.var. Goslin
understands the LIPID MAPS shorthand and the common vendor dialects, so
ether lipids (PE-O, PE-P) and sphingolipids parse correctly.
adata = ov.metabol.annotate_lipids(adata)
print('lipid classes:')
print(adata.var['lipid_class'].value_counts())
lipid classes:
lipid_class
PE 25
PI 22
PE-O 18
PE-P 18
PG 11
PI-P 1
Name: count, dtype: int64
5 — Normalize with PQN#
normalize_pqn applies Probabilistic Quotient Normalization: each
sample is scaled by the median quotient of its lipids against a reference
profile — robust to a few highly variable species. exclude='blank'
drops the solvent blanks before building the reference, and log=True
returns a log2 matrix ready for linear modelling.
norm = ov.metabol.normalize_pqn(adata, measure='Area', exclude='blank', log=True)
print('normalized:', norm.shape, '(blanks dropped)')
print(f'log2 intensity range: {norm.X.min():.1f} .. {norm.X.max():.1f}')
normalized: (56, 101) (blanks dropped)
log2 intensity range: 5.7 .. 23.7
6 — Differential analysis#
ov.metabol.de_lipids runs limma moderated-t — the small-sample
workhorse that borrows variance across lipids, far more powerful than a
per-lipid t-test at n ≈ 11/group. We contrast a high-fat vs normal diet
(both on water). The result table carries lipid-class annotations, which
lsea needs downstream.
de = ov.metabol.de_lipids(
norm, 'HighFat_water - NormalDiet_water', group_col='group',
)
n_sig = int((de['adj.P.Val'] < 0.05).sum())
print(f'{n_sig} / {len(de)} lipids differential at adj.P < 0.05')
de.sort_values('P.Value').head()[
['Molecule', 'Class', 'logFC', 'P.Value', 'adj.P.Val']]
79 / 101 lipids differential at adj.P < 0.05
| Molecule | Class | logFC | P.Value | adj.P.Val | |
|---|---|---|---|---|---|
| 0 | PE(P-38:3) | PE | 1.699273 | 8.614843e-16 | 8.700992e-14 |
| 1 | PE(O-38:4) | PE | 1.765957 | 3.418962e-15 | 1.726576e-13 |
| 2 | PI 34:2 | PI | -1.252535 | 1.469847e-14 | 4.948485e-13 |
| 3 | PG 18:2/18:0 | PG | 1.855818 | 4.051081e-14 | 1.022898e-12 |
| 4 | PG 18:2/18:1 | PG | 2.271831 | 6.715486e-14 | 1.356528e-12 |
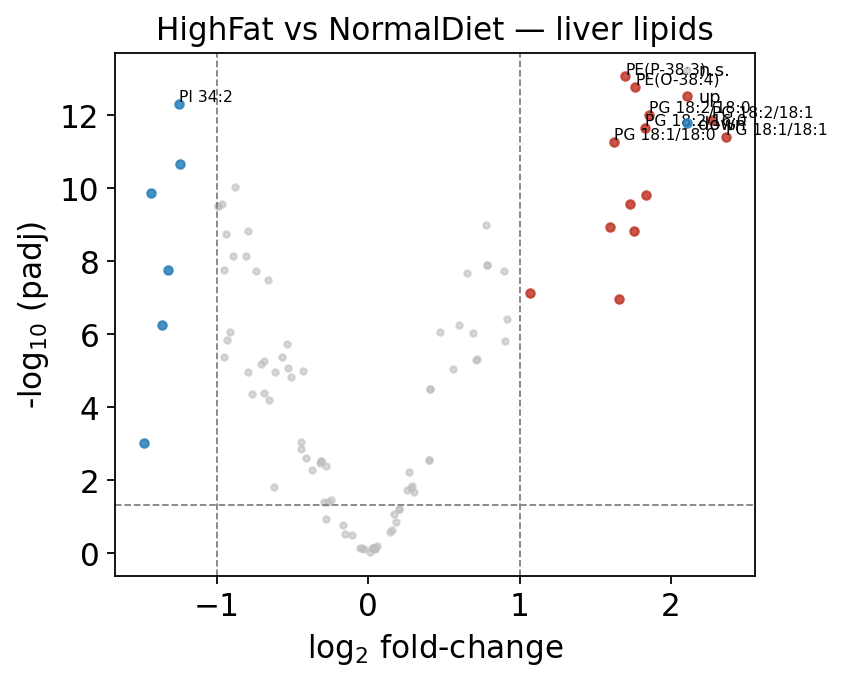
A diet swap remodels the liver lipidome wholesale, so most lipids move —
exactly the regime moderated-t was built for. The volcano below uses
ov.metabol.volcano; we rename the lipidr-style columns to the generic
log2fc / pvalue / padj that omicverse plotting expects.
deg = de.rename(columns={
'logFC': 'log2fc', 'P.Value': 'pvalue', 'adj.P.Val': 'padj',
}).set_index('Molecule')
fig, ax = ov.metabol.volcano(deg, padj_thresh=0.05, log2fc_thresh=1.0,
label_top_n=8)
ax.set_title('HighFat vs NormalDiet — liver lipids')
plt.tight_layout(); plt.show()
7 — Lipid Set Enrichment Analysis#
ov.metabol.lsea runs a preranked GSEA over lipid sets built
automatically from class, total chain length and total unsaturation —
it answers “which lipid groups move coherently?” rather than testing
species one by one. Sets are ranked by enrichment score (ES).
enr = ov.metabol.lsea(de, rank_by='logFC', nperm=2000)
enr.sort_values('pval').head(10)[
['set', 'ES', 'NES', 'pval', 'padj', 'size']]
| set | ES | NES | pval | padj | size | |
|---|---|---|---|---|---|---|
| 0 | Class_PG | 0.966667 | 2.381726 | 0.000846 | 0.015228 | 11 |
| 1 | Class_PI | -0.619609 | -2.047305 | 0.003012 | 0.027108 | 23 |
| 2 | total_cs_4 | 0.705023 | 1.714467 | 0.012584 | 0.075503 | 10 |
| 3 | total_cl_34 | -0.489244 | -1.525266 | 0.040059 | 0.180267 | 19 |
| 4 | total_cl_36 | 0.476344 | 1.450170 | 0.050439 | 0.181579 | 29 |
| 5 | total_cl_32 | -0.593723 | -1.429414 | 0.093516 | 0.246663 | 8 |
| 6 | total_cl_18 | 0.868687 | 1.326131 | 0.108213 | 0.246663 | 2 |
| 7 | Class_SPH | 0.868687 | 1.325317 | 0.109628 | 0.246663 | 2 |
| 8 | total_cs_2 | -0.358807 | -1.123383 | 0.273115 | 0.546230 | 18 |
| 9 | total_cs_3 | -0.359576 | -1.049363 | 0.377868 | 0.672516 | 14 |
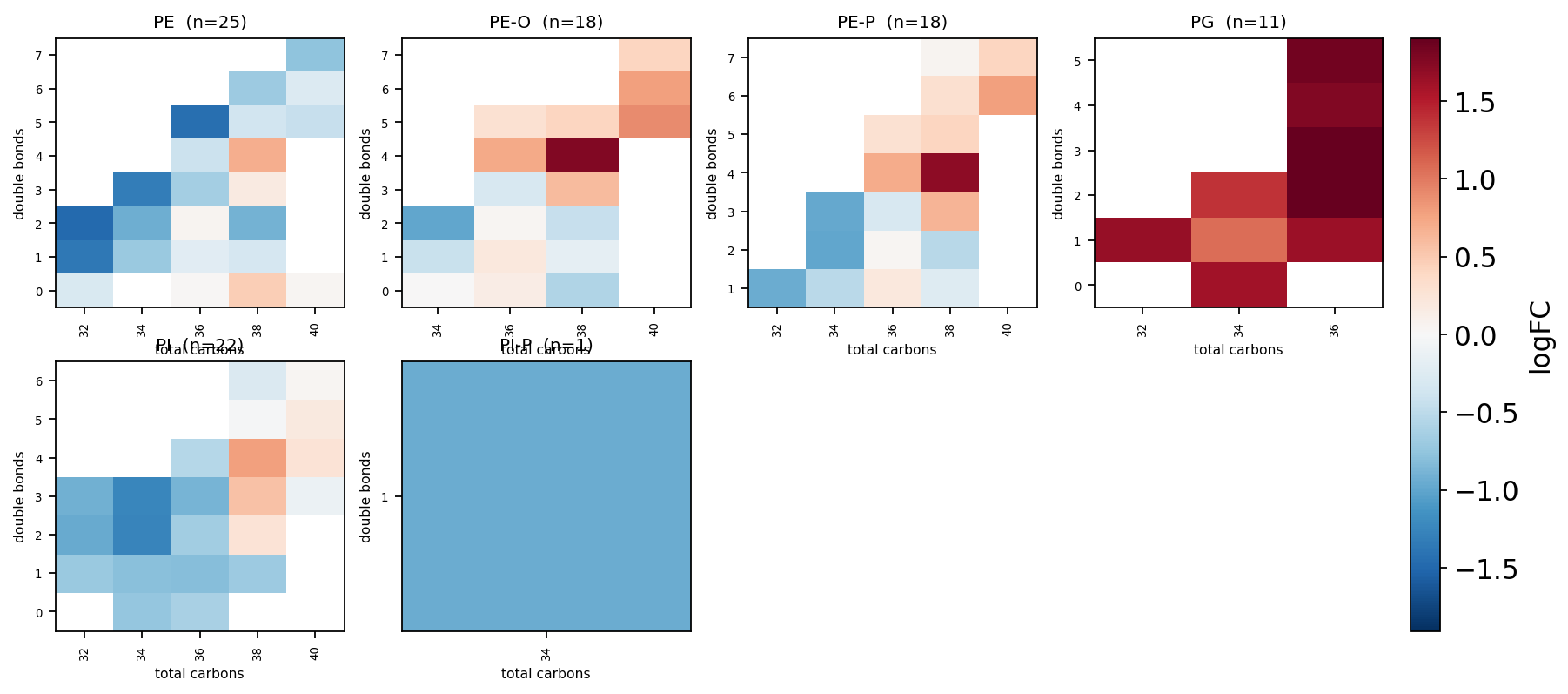
8 — Acyl-chain map#
The signature lipidomics figure: a carbon × double-bond grid per
class, each cell coloured by the differential statistic. It exposes
trends a volcano hides — e.g. whether a class shifts toward longer or
more unsaturated chains. ov.metabol.acyl_chain_map parses every lipid
name and lays out one heatmap panel per class.
fig = ov.metabol.acyl_chain_map(
de.set_index('Molecule'), value_col='logFC', n_cols=4,
)
plt.show()
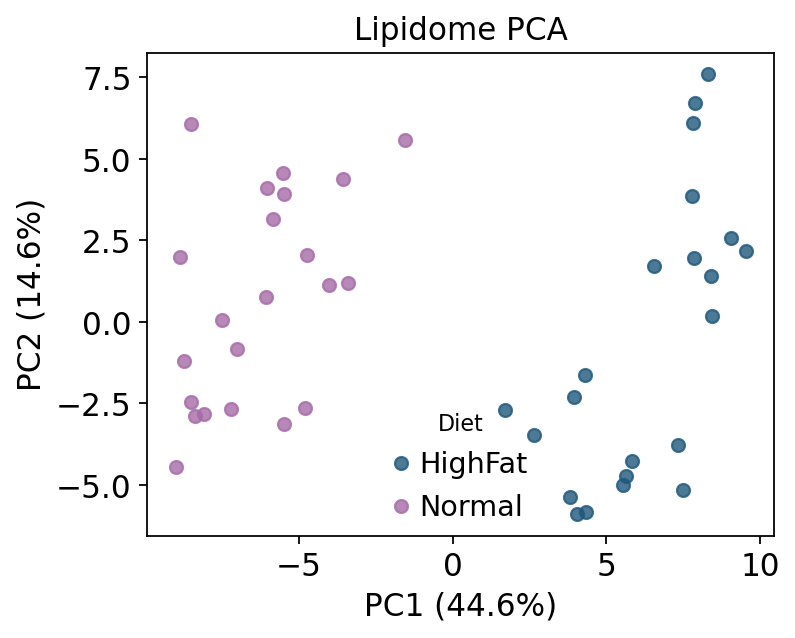
9 — Multivariate analysis#
ov.metabol.lipid_mva provides PCA / PCoA / OPLS-DA on the lipid
matrix. PCA on the biological samples (QC dropped) shows how strongly the
two diets separate. The score plot below colours samples by Diet.
bio = norm[norm.obs['group'] != 'QC'].copy()
mva = ov.metabol.lipid_mva(bio, method='PCA', group_col='Diet')
print('explained variance:', np.round(mva.explained_variance[:3], 3))
explained variance: [0.446 0.146 0.102]
scores = mva.scores
diet = bio.obs['Diet'].reindex(scores.index).astype(str)
fig, ax = plt.subplots(figsize=(5, 4.2))
for g in sorted(diet.unique()):
m = (diet == g).to_numpy()
ax.scatter(scores.iloc[m, 0], scores.iloc[m, 1], label=g, s=34, alpha=0.8)
ax.set_xlabel(f'PC1 ({mva.explained_variance[0] * 100:.1f}%)')
ax.set_ylabel(f'PC2 ({mva.explained_variance[1] * 100:.1f}%)')
ax.legend(title='Diet', frameon=False); ax.set_title('Lipidome PCA')
plt.tight_layout(); plt.show()
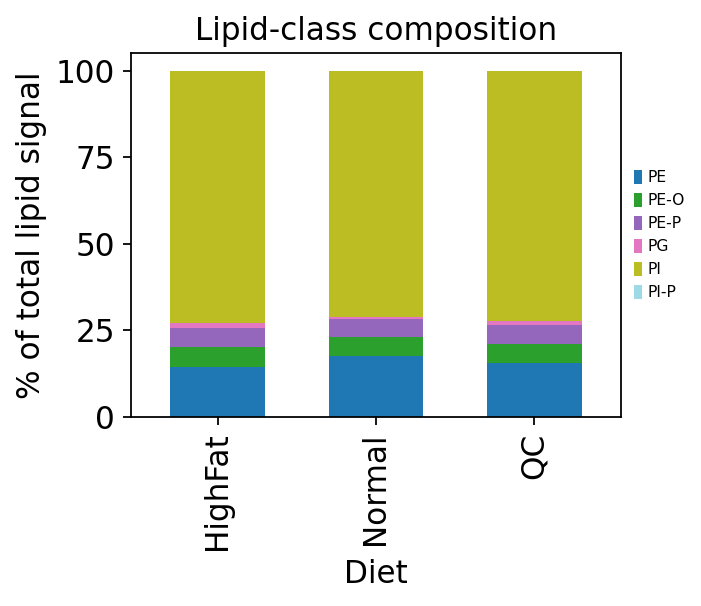
10 — Class-level composition#
For a quick whole-lipidome overview, ov.metabol.aggregate_by_class
collapses the species matrix to per-class totals. A stacked bar of the
mean composition per group is the first plot most lipidomics papers show.
no_blank = adata[adata.obs['group'] != 'blank']
cls = ov.metabol.aggregate_by_class(no_blank, agg='sum')
cls.obs = no_blank.obs
print('class-level matrix:', cls.shape)
cls.var
class-level matrix: (56, 6)
| n_species | |
|---|---|
| PE | 25 |
| PE-O | 18 |
| PE-P | 18 |
| PG | 11 |
| PI | 22 |
| PI-P | 1 |
comp = pd.DataFrame(cls.X, index=cls.obs_names, columns=cls.var_names)
comp['Diet'] = cls.obs['Diet'].values
pct = comp.groupby('Diet').mean().T
pct = pct.div(pct.sum(axis=0), axis=1) * 100
fig, ax = plt.subplots(figsize=(4.6, 4))
pct.T.plot(kind='bar', stacked=True, ax=ax, colormap='tab20', width=0.6)
ax.set_ylabel('% of total lipid signal'); ax.set_xlabel('Diet')
ax.legend(loc='center left', bbox_to_anchor=(1.01, 0.5), frameon=False,
fontsize=7)
ax.set_title('Lipid-class composition')
plt.tight_layout(); plt.show()
Summary#
A complete lipidomics analysis, all AnnData-native:
Step |
|
What it does |
|---|---|---|
Import |
|
Skyline export → AnnData |
Metadata |
|
join clinical table |
Collapse |
|
transitions → one value per lipid |
Annotate |
|
Goslin class / category / chains |
Normalize |
|
PQN / internal-standard |
Differential |
|
limma moderated-t |
Enrichment |
|
lipid-set GSEA · LION ORA |
Multivariate |
|
PCA / PCoA / OPLS-DA |
Visualize |
|
The analysis engine is the standalone, R-parity-tested pylipidr port of
Bioconductor lipidr; ov.metabol exposes it as registered functions so
lipidomics composes with every other omicverse module.